Division of Infection and Immunity, The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, 3052, Australia.
Department of Medical Biology, The University of Melbourne, Parkville, VIC, 3010, Australia.
Cell Death Differ. 2018 May;25(5):951-965. doi: 10.1038/s41418-017-0031-1. Epub 2017 Dec 11.
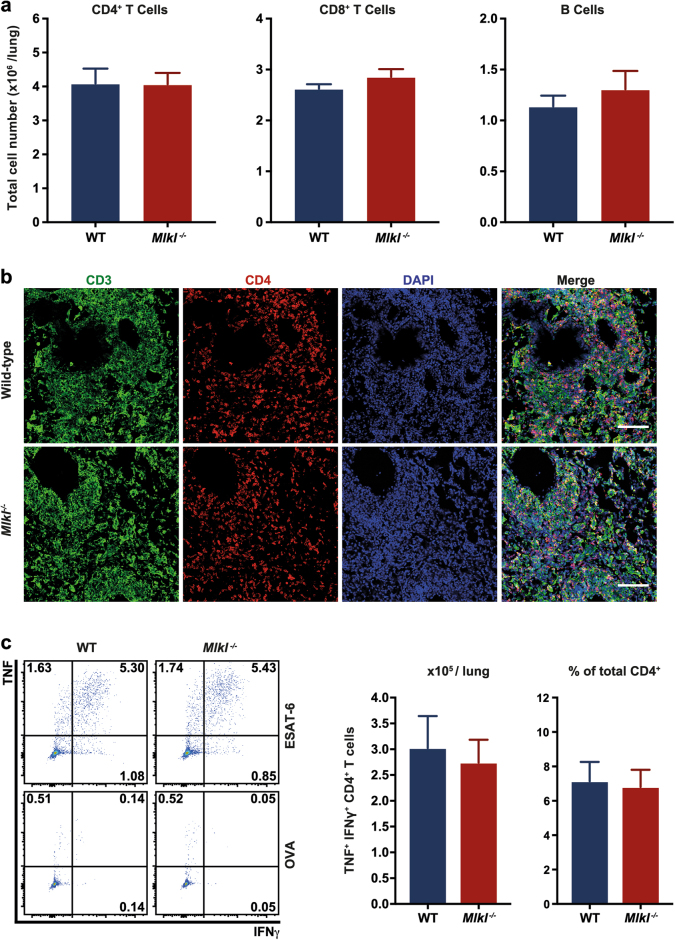
Mixed lineage kinase domain-like (MLKL)-dependent necroptosis is thought to be implicated in the death of mycobacteria-infected macrophages, reportedly allowing escape and dissemination of the microorganism. Given the consequent interest in developing inhibitors of necroptosis to treat Mycobacterium tuberculosis (Mtb) infection, we used human pharmacologic and murine genetic models to definitively establish the pathophysiological role of necroptosis in Mtb infection. We observed that Mtb infection of macrophages remodeled the intracellular signaling landscape by upregulating MLKL, TNFR1, and ZBP1, whilst downregulating cIAP1, thereby establishing a strong pro-necroptotic milieu. However, blocking necroptosis either by deleting Mlkl or inhibiting RIPK1 had no effect on the survival of infected human or murine macrophages. Consistent with this, MLKL-deficiency or treatment of humanized mice with the RIPK1 inhibitor Nec-1s did not impact on disease outcomes in vivo, with mice displaying lung histopathology and bacterial burdens indistinguishable from controls. Therefore, although the necroptotic pathway is primed by Mtb infection, macrophage necroptosis is ultimately restricted to mitigate disease pathogenesis. We identified cFLIP upregulation that may promote caspase 8-mediated degradation of CYLD, and other necrosome components, as a possible mechanism abrogating Mtb's capacity to coopt necroptotic signaling. Variability in the capacity of these mechanisms to interfere with necroptosis may influence disease severity and could explain the heterogeneity of Mtb infection and disease.
混合谱系激酶结构域样(MLKL)依赖性坏死被认为与分枝杆菌感染的巨噬细胞死亡有关,据报道这允许微生物的逃逸和传播。鉴于人们对开发坏死抑制剂来治疗结核分枝杆菌(Mtb)感染的浓厚兴趣,我们使用了人类药理学和小鼠遗传学模型来明确确定坏死在 Mtb 感染中的病理生理作用。我们观察到 Mtb 感染巨噬细胞通过上调 MLKL、TNFR1 和 ZBP1,同时下调 cIAP1,重塑细胞内信号景观,从而建立了强烈的促坏死环境。然而,通过删除 Mlkl 或抑制 RIPK1 来阻断坏死对感染的人或鼠巨噬细胞的存活没有影响。与此一致的是,MLKL 缺陷或用 RIPK1 抑制剂 Nec-1s 治疗人源化小鼠对体内疾病结果没有影响,小鼠显示的肺组织病理学和细菌负荷与对照无区别。因此,尽管坏死途径被 Mtb 感染引发,但巨噬细胞坏死最终受到限制,以减轻疾病发病机制。我们发现 cFLIP 的上调可能促进 caspase 8 介导的 CYLD 和其他坏死体成分的降解,这可能是一种阻止 Mtb 利用坏死信号的机制。这些机制干扰坏死的能力的可变性可能影响疾病的严重程度,并可以解释 Mtb 感染和疾病的异质性。