Ruiz-Arenas Carlos, González Juan R
ISGlobal, Centre for Research in Environmental Epidemiology (CREAL), Barcelona, Spain.
Universitat Pompeu Fabra (UPF), Barcelona, Spain.
BMC Bioinformatics. 2017 Dec 14;18(1):553. doi: 10.1186/s12859-017-1986-0.
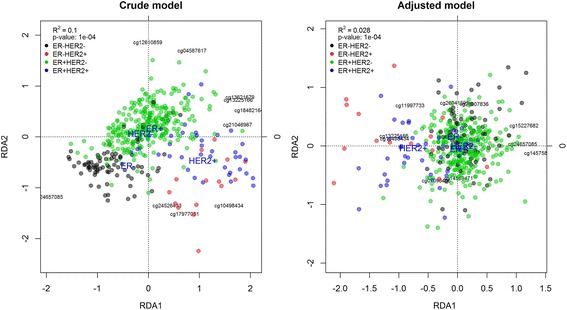
DNA methylation is an epigenetic process that regulates gene expression. Methylation can be modified by environmental exposures and changes in the methylation patterns have been associated with diseases. Methylation microarrays measure methylation levels at more than 450,000 CpGs in a single experiment, and the most common analysis strategy is to perform a single probe analysis to find methylation probes associated with the outcome of interest. However, methylation changes usually occur at the regional level: for example, genomic structural variants can affect methylation patterns in regions up to several megabases in length. Existing DMR methods provide lists of Differentially Methylated Regions (DMRs) of up to only few kilobases in length, and cannot check if a target region is differentially methylated. Therefore, these methods are not suitable to evaluate methylation changes in large regions. To address these limitations, we developed a new DMR approach based on redundancy analysis (RDA) that assesses whether a target region is differentially methylated.
Using simulated and real datasets, we compared our approach to three common DMR detection methods (Bumphunter, blockFinder, and DMRcate). We found that Bumphunter underestimated methylation changes and blockFinder showed poor performance. DMRcate showed poor power in the simulated datasets and low specificity in the real data analysis. Our method showed very high performance in all simulation settings, even with small sample sizes and subtle methylation changes, while controlling type I error. Other advantages of our method are: 1) it estimates the degree of association between the DMR and the outcome; 2) it can analyze a targeted or region of interest; and 3) it can evaluate the simultaneous effects of different variables. The proposed methodology is implemented in MEAL, a Bioconductor package designed to facilitate the analysis of methylation data.
We propose a multivariate approach to decipher whether an outcome of interest alters the methylation pattern of a region of interest. The method is designed to analyze large target genomic regions and outperforms the three most popular methods for detecting DMRs. Our method can evaluate factors with more than two levels or the simultaneous effect of more than one continuous variable, which is not possible with the state-of-the-art methods.
DNA甲基化是一种调节基因表达的表观遗传过程。甲基化可被环境暴露所修饰,且甲基化模式的改变与疾病相关。甲基化微阵列在单次实验中可测量超过45万个CpG位点的甲基化水平,最常见的分析策略是进行单探针分析以找到与感兴趣结果相关的甲基化探针。然而,甲基化变化通常发生在区域水平:例如,基因组结构变异可影响长达数兆碱基区域的甲基化模式。现有的差异甲基化区域(DMR)方法提供的DMR列表长度仅达几千碱基,且无法检查目标区域是否存在差异甲基化。因此,这些方法不适用于评估大区域内的甲基化变化。为解决这些局限性,我们基于冗余分析(RDA)开发了一种新的DMR方法,该方法可评估目标区域是否存在差异甲基化。
使用模拟数据集和真实数据集,我们将我们的方法与三种常见的DMR检测方法(Bumphunter、blockFinder和DMRcate)进行了比较。我们发现Bumphunter低估了甲基化变化,而blockFinder表现不佳。DMRcate在模拟数据集中功效较差,在真实数据分析中特异性较低。我们的方法在所有模拟设置中均表现出非常高的性能,即使样本量较小且甲基化变化细微,同时还能控制I型错误。我们方法的其他优点包括:1)它可估计DMR与结果之间的关联程度;2)它可分析目标区域或感兴趣区域;3)它可评估不同变量的同时效应。所提出的方法在MEAL中实现,MEAL是一个旨在促进甲基化数据分析的Bioconductor软件包。
我们提出了一种多变量方法来解读感兴趣的结果是否会改变感兴趣区域的甲基化模式。该方法旨在分析大型目标基因组区域,并且优于三种最流行的DMR检测方法。我们的方法可评估具有两个以上水平的因素或多个连续变量的同时效应,这是现有方法无法做到的。