Jergova Stanislava, Gordon Catherine E, Gajavelli Shyam, Sagen Jacqueline
The Miami Project, Miller School of Medicine, University of Miami, Miami, FL, United States.
Front Mol Neurosci. 2017 Dec 8;10:406. doi: 10.3389/fnmol.2017.00406. eCollection 2017.
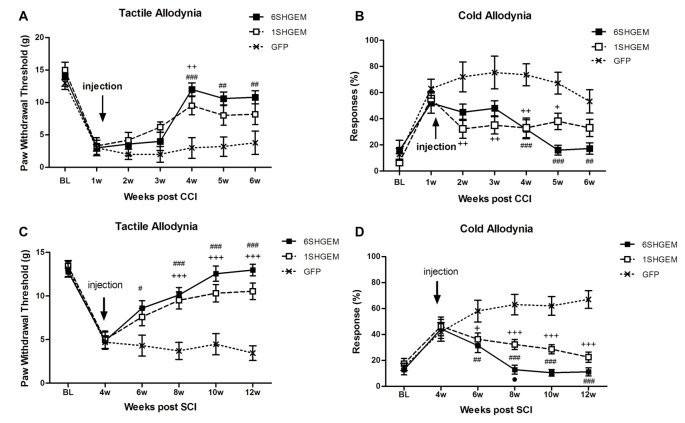
The insufficient pain relief provided by current pharmacotherapy for chronic neuropathic pain is a serious medical problem. The enhanced glutamate signaling via NMDA receptors appears to be one of the key events in the development of chronic pain. Although effective, clinical use of systemic NMDA antagonists is limited by adverse effects such as hallucinations and motor dysfunction. Opioids are also potent analgesics but their chronic use is accompanied by tolerance and risk of addiction. However, combination of NMDA antagonists and opioids seems to provide a stable pain relieve at subthreshold doses of both substances, eliminating development of side effects. Our previous research showed that combined delivery of NMDA antagonist Serine histrogranin (SHG) and endomorphin1 (EM1) leads to attenuation of acute and chronic pain. The aim of this study was to design and evaluate an analgesic potency of the gene construct encoding SHG and EM1. Constructs with 1SHG copy in combination with EM1, 1SHG/EM1, and 6SHG/EM1 were intraspinally injected to animals with peripheral nerve injury-induced pain (chronic constriction injury, CCI) or spinal cord injury induced pain (clip compression model, SCI) and tactile and cold allodynia were evaluated. AAV2/8 particles were used for gene delivery. The results demonstrated 6SHG/EM1 as the most efficient for alleviation of pain-related behavior. The effect was observed up to 8 weeks in SCI animals, suggesting the lack of tolerance of possible synergistic effect between SHG and EM1. Intrathecal injection of SHG antibody or naloxone attenuated the analgesic effect in treated animals. Biochemical and histochemical evaluation confirmed the presence of both peptides in the spinal tissue. The results of this study showed that the injection of AAV vectors encoding combined SHG/EM constructs can provide long term attenuation of pain without overt adverse side effects. This approach may provide better treatment options for patients suffering from chronic pain.
目前用于治疗慢性神经性疼痛的药物疗法所提供的疼痛缓解不足是一个严重的医学问题。通过N-甲基-D-天冬氨酸(NMDA)受体增强的谷氨酸信号传导似乎是慢性疼痛发展中的关键事件之一。尽管全身使用NMDA拮抗剂有效,但其临床应用受到诸如幻觉和运动功能障碍等不良反应的限制。阿片类药物也是强效镇痛药,但其长期使用会伴随着耐受性和成瘾风险。然而,NMDA拮抗剂和阿片类药物联合使用似乎在两种物质的阈下剂量时能提供稳定的疼痛缓解,且能消除副作用的产生。我们之前的研究表明,联合递送NMDA拮抗剂丝氨酸组蛋白聚糖(SHG)和内吗啡肽1(EM1)可减轻急性和慢性疼痛。本研究的目的是设计并评估编码SHG和EM1的基因构建体的镇痛效力。将含有1个SHG拷贝与EM1组合的构建体(1SHG/EM1)以及6SHG/EM1构建体经脊髓内注射到患有外周神经损伤诱导性疼痛(慢性缩窄损伤,CCI)或脊髓损伤诱导性疼痛(夹压模型,SCI)的动物体内,并评估触觉和冷觉异常性疼痛。使用腺相关病毒2/8(AAV2/8)颗粒进行基因递送。结果表明,6SHG/EM1在减轻疼痛相关行为方面最为有效。在SCI动物中,这种效果可持续长达8周,表明SHG和EM1之间可能存在的协同作用不存在耐受性。鞘内注射SHG抗体或纳洛酮可减弱治疗动物的镇痛效果。生化和组织化学评估证实脊髓组织中存在这两种肽。本研究结果表明,注射编码SHG/EM联合构建体的腺相关病毒载体可长期减轻疼痛,且无明显不良副作用。这种方法可能为慢性疼痛患者提供更好的治疗选择。