Wellcome Trust Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridgeshire, CB10 1SA, UK.
Department of Medicine, University of Cambridge, Box 157, Addenbrooke's Hospital, Hills Road, Cambridge, CB2 0QQ, UK.
Genome Med. 2017 Dec 27;9(1):119. doi: 10.1186/s13073-017-0507-0.
Enterococcus faecium is a leading cause of hospital-acquired infection, particularly in the immunocompromised. Here, we use whole genome sequencing of E. faecium to study within-host evolution and the transition from gut carriage to invasive disease.
We isolated and sequenced 180 E. faecium from four immunocompromised patients who developed bloodstream infection during longitudinal surveillance of E. faecium in stool and their immediate environment.
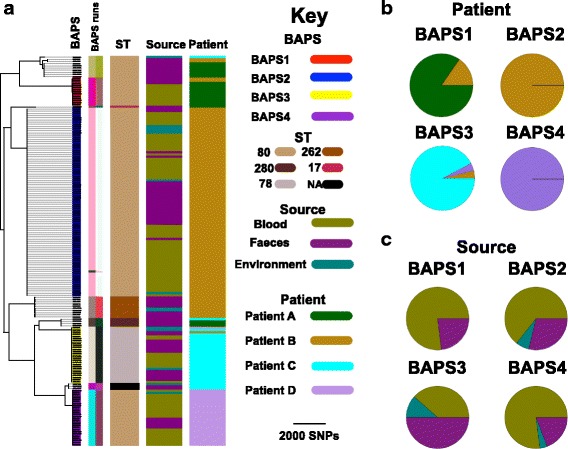
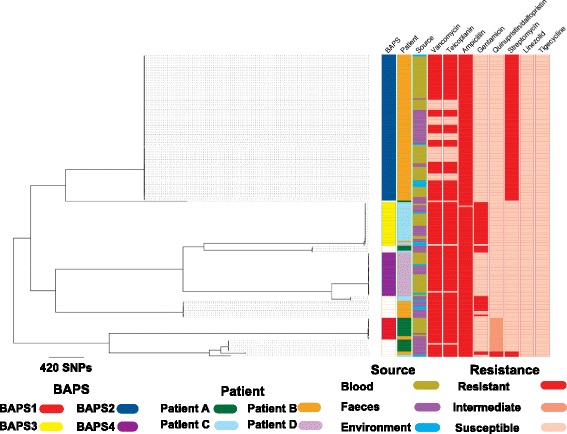
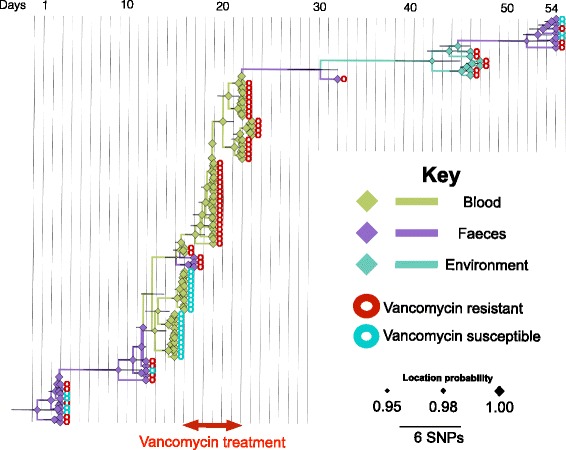
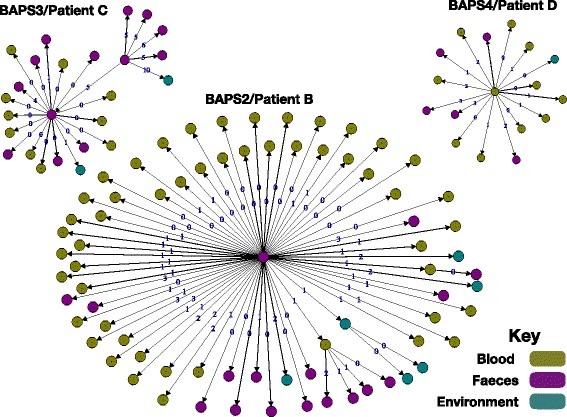
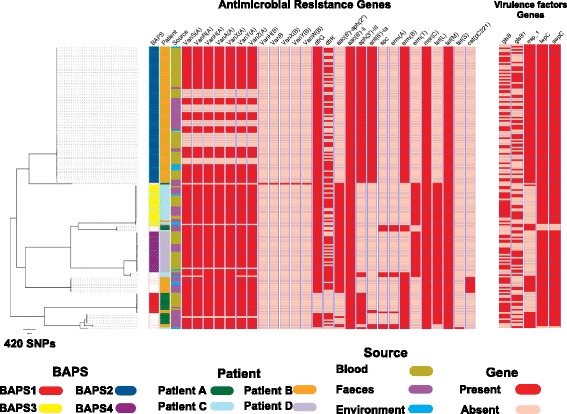
A phylogenetic tree based on single nucleotide polymorphisms (SNPs) in the core genome of the 180 isolates demonstrated several distinct clones. This was highly concordant with the population structure inferred by Bayesian methods, which contained four main BAPS (Bayesian Analysis of Population Structure) groups. The majority of isolates from each patient resided in a single group, but all four patients also carried minority populations in stool from multiple phylogenetic groups. Bloodstream isolates from each case belonged to a single BAPS group, which differed in all four patients. Analysis of 87 isolates (56 from blood) belonging to a single BAPS group that were cultured from the same patient over 54 days identified 30 SNPs in the core genome (nine intergenic, 13 non-synonymous, eight synonymous), and 250 accessory genes that were variably present. Comparison of these genetic variants in blood isolates versus those from stool or environment did not identify any variants associated with bloodstream infection. The substitution rate for these isolates was estimated to be 128 (95% confidence interval 79.82 181.77) mutations per genome per year, more than ten times higher than previous estimates for E. faecium. Within-patient variation in vancomycin resistance associated with vanA was common and could be explained by plasmid loss, or less often by transposon loss.
These findings demonstrate the diversity of E. faecium carriage by individual patients and significant within-host diversity of E. faecium, but do not provide evidence for adaptive genetic variation associated with invasion.
屎肠球菌是医院获得性感染的主要原因,尤其是在免疫功能低下的人群中。在这里,我们使用屎肠球菌的全基因组测序来研究宿主内的进化以及从肠道携带到侵袭性疾病的转变。
我们从 4 名免疫功能低下的患者中分离并测序了 180 株屎肠球菌,这些患者在对粪便和其周围环境中的屎肠球菌进行纵向监测期间发生了血流感染。
基于 180 株分离株核心基因组中的单核苷酸多态性(SNP)构建的系统发育树显示了几个不同的克隆。这与贝叶斯方法推断的种群结构高度一致,该方法包含四个主要的 BAPS(贝叶斯种群结构分析)组。每个患者的大多数分离株都位于一个单一的组中,但所有四个患者的粪便中还携带来自多个系统发育群的少数群体。每个病例的血流分离株都属于一个单一的 BAPS 组,在所有四个患者中都不同。对来自同一患者的 54 天内培养的属于单一 BAPS 组的 87 株(56 株来自血液)进行分析,在核心基因组中发现了 30 个 SNP(9 个基因间,13 个非同义,8 个同义)和 250 个可变异的辅助基因。将这些血液分离株的遗传变异与粪便或环境中的分离株进行比较,未发现与血流感染相关的任何变异。这些分离株的取代率估计为每个基因组每年 128(95%置信区间 79.82-181.77)个突变,比以前估计的屎肠球菌要高 10 多倍。与万古霉素抗性相关的 vanA 基因的患者内变异性很常见,这可以通过质粒丢失来解释,或者不太常见的是通过转座子丢失来解释。
这些发现表明了个体患者屎肠球菌携带的多样性以及屎肠球菌在宿主内的显著多样性,但没有提供与侵袭相关的适应性遗传变异的证据。