Division of Psychiatry, University of Edinburgh, Royal Edinburgh Hospital, EH10 5HF, Edinburgh, UK.
Institute of Genetic Medicine, Newcastle University, NE1 7RU, Newcastle upon Tyne, UK.
Transl Psychiatry. 2018 Jan 10;8(1):9. doi: 10.1038/s41398-017-0034-1.
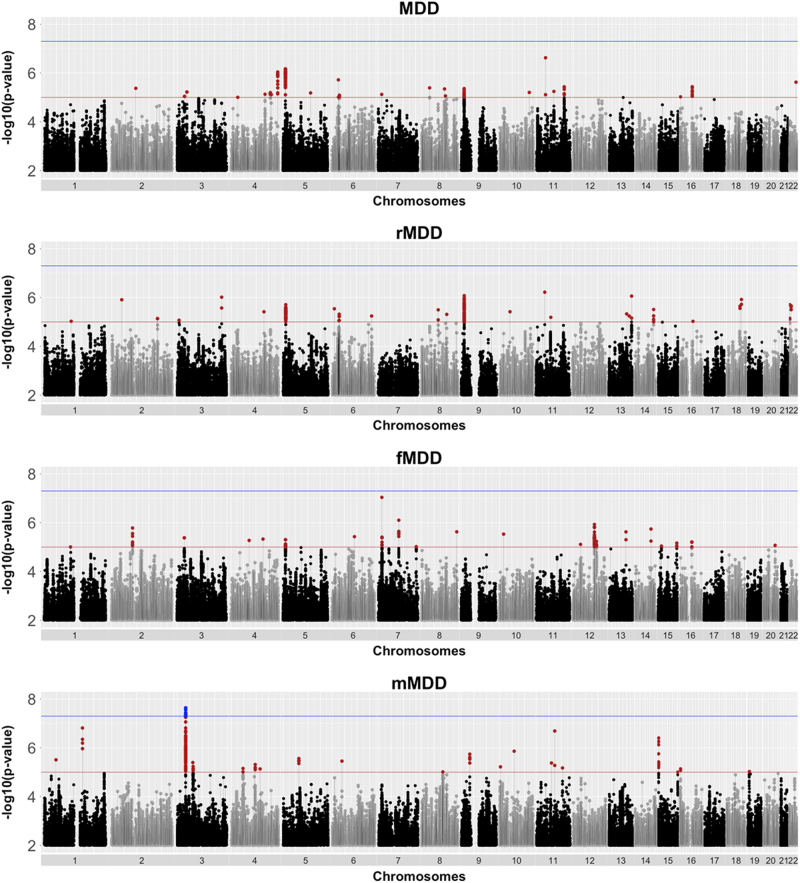
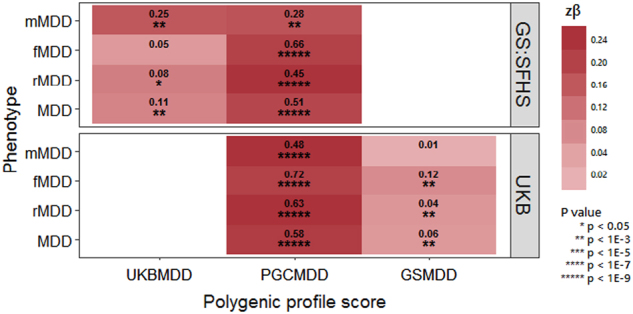
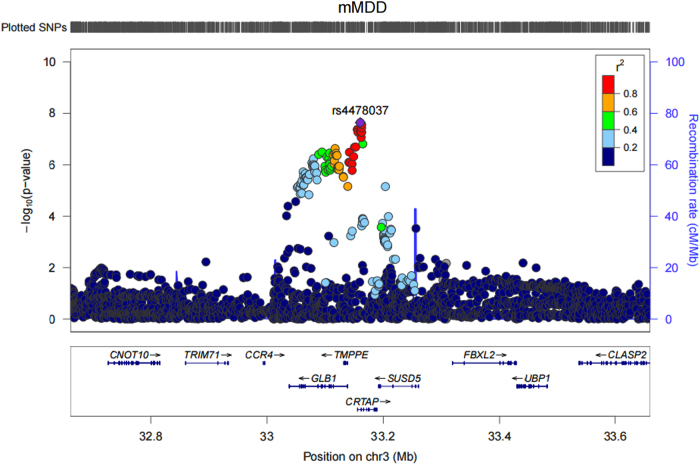
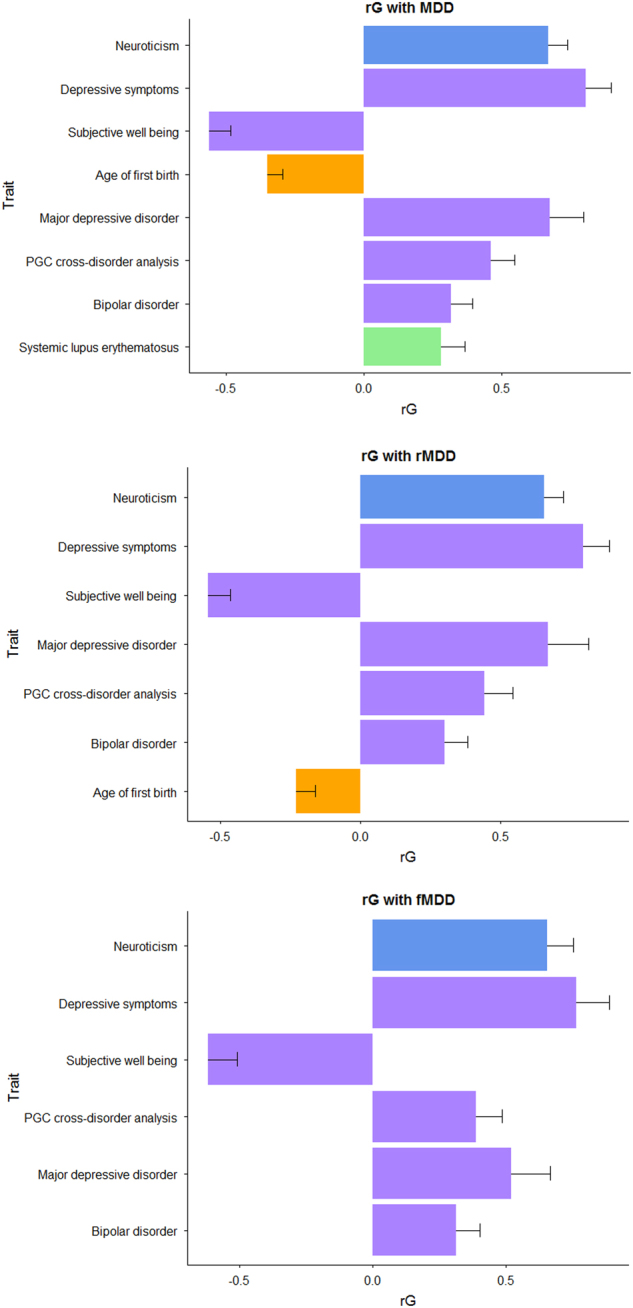
Few replicable genetic associations for Major Depressive Disorder (MDD) have been identified. Recent studies of MDD have identified common risk variants by using a broader phenotype definition in very large samples, or by reducing phenotypic and ancestral heterogeneity. We sought to ascertain whether it is more informative to maximize the sample size using data from all available cases and controls, or to use a sex or recurrent stratified subset of affected individuals. To test this, we compared heritability estimates, genetic correlation with other traits, variance explained by MDD polygenic score, and variants identified by genome-wide meta-analysis for broad and narrow MDD classifications in two large British cohorts - Generation Scotland and UK Biobank. Genome-wide meta-analysis of MDD in males yielded one genome-wide significant locus on 3p22.3, with three genes in this region (CRTAP, GLB1, and TMPPE) demonstrating a significant association in gene-based tests. Meta-analyzed MDD, recurrent MDD and female MDD yielded equivalent heritability estimates, showed no detectable difference in association with polygenic scores, and were each genetically correlated with six health-correlated traits (neuroticism, depressive symptoms, subjective well-being, MDD, a cross-disorder phenotype and Bipolar Disorder). Whilst stratified GWAS analysis revealed a genome-wide significant locus for male MDD, the lack of independent replication, and the consistent pattern of results in other MDD classifications suggests that phenotypic stratification using recurrence or sex in currently available sample sizes is currently weakly justified. Based upon existing studies and our findings, the strategy of maximizing sample sizes is likely to provide the greater gain.
目前已经确定了少数可复制的与重度抑郁症(MDD)相关的遗传关联。最近的 MDD 研究通过在非常大的样本中使用更广泛的表型定义或通过减少表型和祖先异质性来确定常见的风险变异。我们试图确定通过使用所有可用病例和对照的数据来最大化样本量是否更具信息量,或者使用受影响个体的性别或复发性分层子集。为了测试这一点,我们比较了两个大型英国队列 - 苏格兰世代和英国生物库中广泛和狭义 MDD 分类的遗传率估计、与其他特征的遗传相关性、MDD 多基因评分解释的方差以及全基因组荟萃分析确定的变体。对男性 MDD 的全基因组荟萃分析产生了一个位于 3p22.3 上的全基因组显著位点,该区域的三个基因(CRTAP、GLB1 和 TMPPE)在基因测试中显示出显著的关联。荟萃分析的 MDD、复发性 MDD 和女性 MDD 产生了等效的遗传率估计值,与多基因评分的关联没有检测到差异,并且与六个与健康相关的特征(神经质、抑郁症状、主观幸福感、MDD、跨疾病表型和双相情感障碍)均具有遗传相关性。虽然分层 GWAS 分析揭示了男性 MDD 的全基因组显著位点,但缺乏独立的复制,以及其他 MDD 分类中一致的结果模式表明,目前在可用样本量中使用复发或性别进行表型分层目前依据不足。基于现有研究和我们的发现,最大化样本量的策略可能会带来更大的收益。