Mashima Ryuichi, Ohira Mari, Okuyama Torayuki, Tatsumi Akiya
Department of Clinical Laboratory Medicine, National Center for Child Health and Development, 2-10-1 Okura, Setagaya-ku, Tokyo 157-8535, Japan.
Mol Genet Metab Rep. 2017 Dec 21;14:36-40. doi: 10.1016/j.ymgmr.2017.12.001. eCollection 2018 Mar.
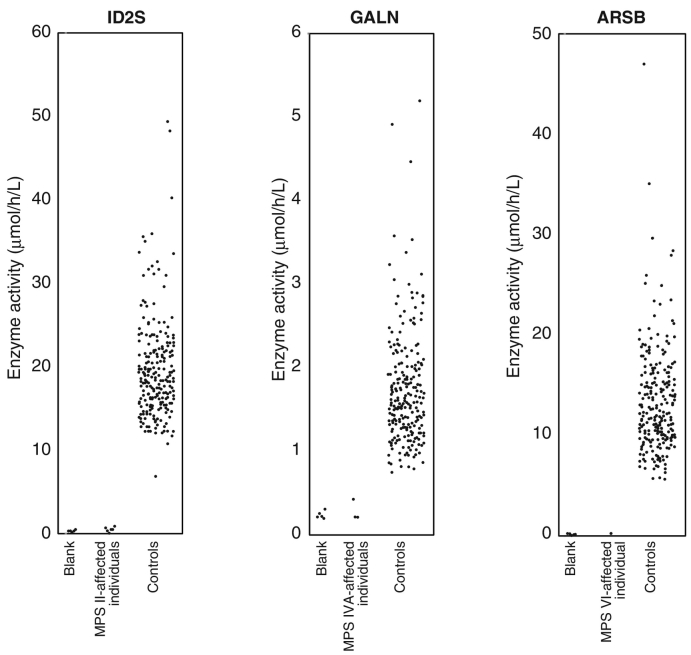
Mucopolysaccharidosis (MPS) is a genetic disorder characterized by the accumulation of glycosaminoglycans in the body. Of the multiple MPS disease subtypes, several are caused by defects in sulfatases. Specifically, a defect in iduronate-2-sulfatase (ID2S) leads to MPS II, whereas -acetylgalactosamine-6-sulfatase (GALN) and -acetylgalactosamine-4-sulfatase (ARSB) defects relate to MPS IVA and MPS VI, respectively. A previous study reported a combined assay for these three disorders in a 96-well plate using a liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based technique (Kumar et al., Clin Chem 2015 61(11):1363-1371). In our study, we applied this methodology to a Japanese population to examine the assay precision and the separation of populations between disease-affected individuals and controls for these three disorders. Within our assay conditions, the coefficient of variation (CV, %) values for an interday assay of ID2S, GALN, and ARSB were 9%, 18%, and 9%, respectively ( = 7). The average enzyme activities of ID2S, GALN, and ARSB in random neonates were 19.6 ± 5.8, 1.7 ± 0.7, and 13.4 ± 5.2 μmol/h/L (mean ± SD, = 240), respectively. In contrast, the average enzyme activities of ID2S, GALN, and ARSB in disease-affected individuals were 0.5 ± 0.2 ( = 6), 0.3 ± 0.1 ( = 3), and 0.3 ( = 1) μmol/h/L, respectively. The representative analytical range values corresponding to ID2S, GALN, and ARSB were 39, 17, and 168, respectively. These results raise the possibility that the population of disease-affected individuals could be separated from that of healthy individuals using the LC-MS/MS-based technique.
黏多糖贮积症(MPS)是一种遗传性疾病,其特征是体内糖胺聚糖积累。在多种MPS疾病亚型中,有几种是由硫酸酯酶缺陷引起的。具体而言,艾杜糖醛酸-2-硫酸酯酶(ID2S)缺陷导致MPS II,而N-乙酰半乳糖胺-6-硫酸酯酶(GALN)和N-乙酰半乳糖胺-4-硫酸酯酶(ARSB)缺陷分别与MPS IVA和MPS VI相关。先前的一项研究报告了使用基于液相色谱-串联质谱(LC-MS/MS)技术在96孔板中对这三种疾病进行联合检测的方法(Kumar等人,《临床化学》2015年61(11):1363 - 1371)。在我们的研究中,我们将这种方法应用于日本人群,以检验这三种疾病在疾病患者和对照人群之间检测的精密度以及人群分离情况。在我们的检测条件下,ID2S、GALN和ARSB日间检测的变异系数(CV,%)值分别为9%、18%和9%(n = 7)。随机新生儿中ID2S、GALN和ARSB的平均酶活性分别为19.6±5.8、1.7±0.7和13.4±5.2 μmol/h/L(平均值±标准差,n = 240)。相比之下,疾病患者中ID2S、GALN和ARSB的平均酶活性分别为0.5±0.2(n = 6)、0.3±0.1(n = 3)和0.3(n = 1)μmol/h/L。与ID2S、GALN和ARSB对应的代表性分析范围值分别为39、17和168。这些结果增加了使用基于LC-MS/MS的技术将疾病患者群体与健康个体群体分离的可能性。