IBISC, Univ Evry, Université Paris-Saclay, Evry, 91025, France.
BMC Bioinformatics. 2018 Jan 15;19(1):13. doi: 10.1186/s12859-018-2007-7.
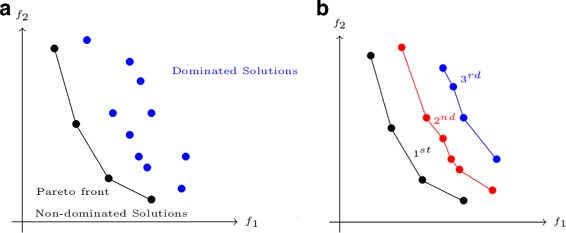
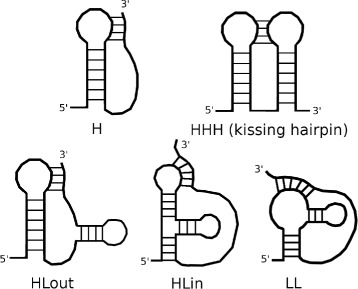
RNA structure prediction is an important field in bioinformatics, and numerous methods and tools have been proposed. Pseudoknots are specific motifs of RNA secondary structures that are difficult to predict. Almost all existing methods are based on a single model and return one solution, often missing the real structure. An alternative approach would be to combine different models and return a (small) set of solutions, maximizing its quality and diversity in order to increase the probability that it contains the real structure.
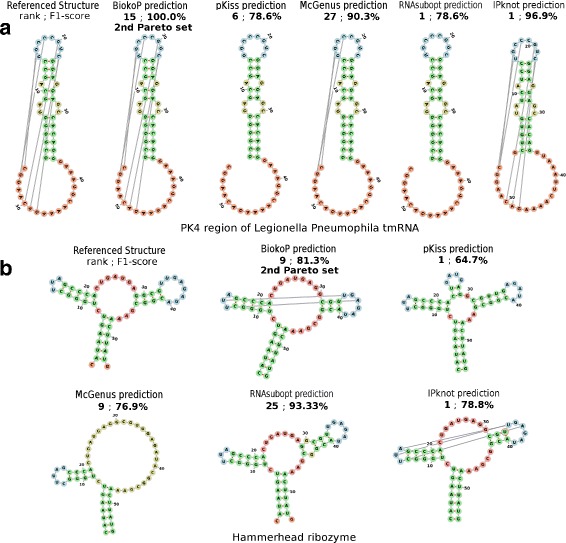
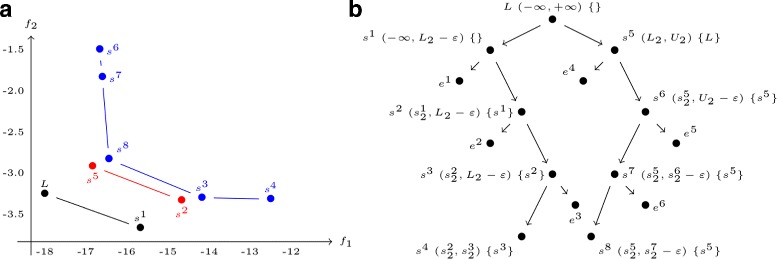
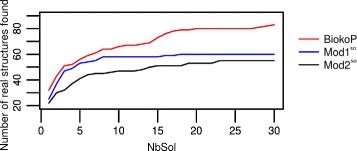
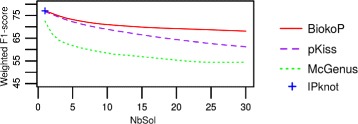
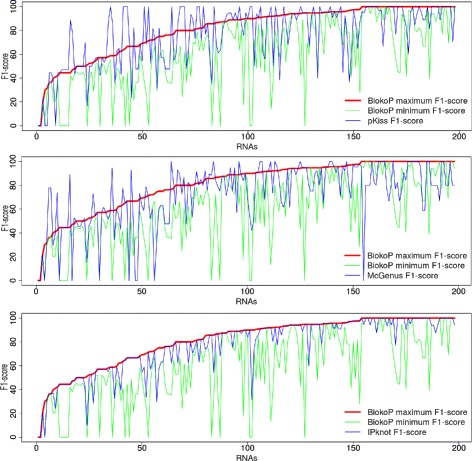
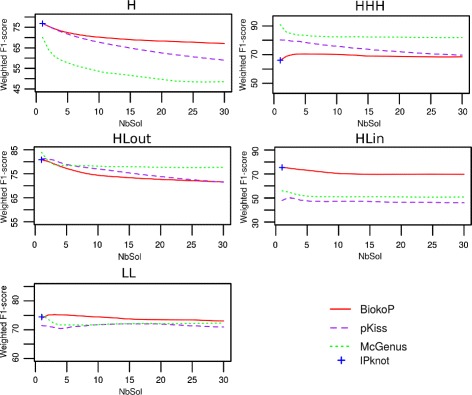
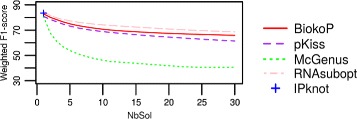
We propose here an original method for predicting RNA secondary structures with pseudoknots, based on integer programming. We developed a generic bi-objective integer programming algorithm allowing to return optimal and sub-optimal solutions optimizing simultaneously two models. This algorithm was then applied to the combination of two known models of RNA secondary structure prediction, namely MEA and MFE. The resulting tool, called BiokoP, is compared with the other methods in the literature. The results show that the best solution (structure with the highest F-score) is, in most cases, given by BiokoP. Moreover, the results of BiokoP are homogeneous, regardless of the pseudoknot type or the presence or not of pseudoknots. Indeed, the F-scores are always higher than 70% for any number of solutions returned.
The results obtained by BiokoP show that combining the MEA and the MFE models, as well as returning several optimal and several sub-optimal solutions, allow to improve the prediction of secondary structures. One perspective of our work is to combine better mono-criterion models, in particular to combine a model based on the comparative approach with the MEA and the MFE models. This leads to develop in the future a new multi-objective algorithm to combine more than two models. BiokoP is available on the EvryRNA platform: https://EvryRNA.ibisc.univ-evry.fr .
RNA 结构预测是生物信息学中的一个重要领域,已经提出了许多方法和工具。假结是 RNA 二级结构的特定模体,难以预测。几乎所有现有的方法都基于单一模型并返回一个解决方案,经常会错过真实结构。另一种方法是结合不同的模型并返回一个(小)解决方案集,以最大化其质量和多样性,从而增加包含真实结构的可能性。
我们在这里提出了一种基于整数规划的预测具有假结的 RNA 二级结构的原始方法。我们开发了一种通用的双目标整数规划算法,可以同时优化两个模型以返回最优和次优解。然后将该算法应用于两种已知的 RNA 二级结构预测模型,即 MEA 和 MFE 的组合。由此产生的工具称为 BiokoP,并与文献中的其他方法进行了比较。结果表明,在大多数情况下,最好的解决方案(具有最高 F 分数的结构)由 BiokoP 给出。此外,BiokoP 的结果是同质的,与假结类型、是否存在假结无关。实际上,对于返回的任何数量的解决方案,F 分数始终高于 70%。
BiokoP 获得的结果表明,结合 MEA 和 MFE 模型以及返回多个最优和多个次优解决方案可以提高二级结构的预测。我们工作的一个前景是结合更好的单准则模型,特别是将基于比较方法的模型与 MEA 和 MFE 模型结合起来。这将导致未来开发一种新的多目标算法来结合两个以上的模型。BiokoP 可在 EvryRNA 平台上获得:https://EvryRNA.ibisc.univ-evry.fr。