Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Via S. Pansini 5, Naples, 80131, Italy.
Department of Advanced Biomedical Sciences, University of Naples Federico II, Via S. Pansini 5, Naples, 80131, Italy.
Cell Death Dis. 2018 Jan 18;9(2):40. doi: 10.1038/s41419-017-0187-0.
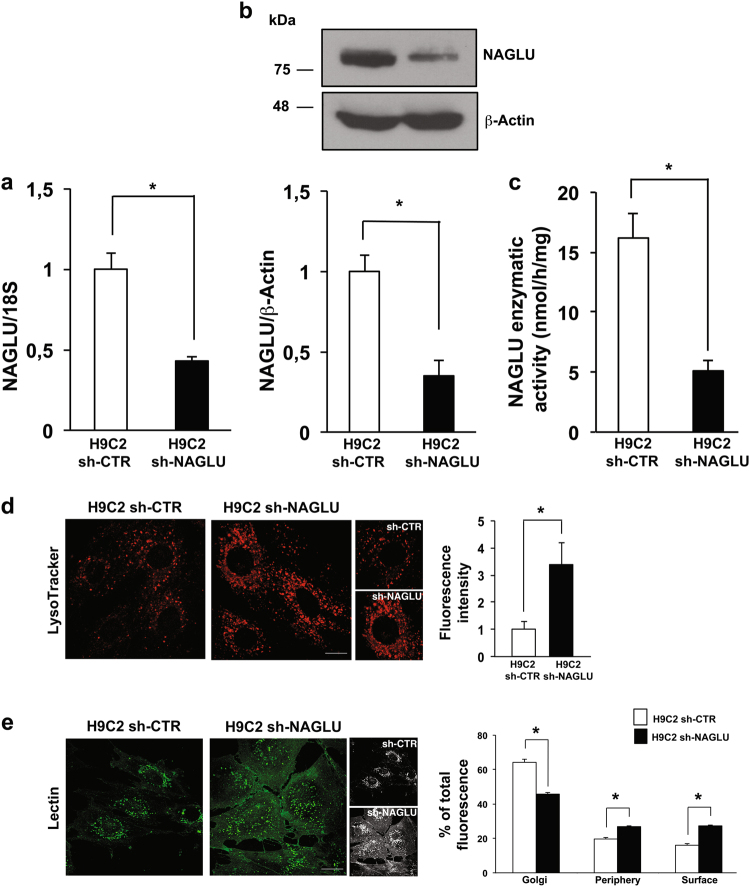
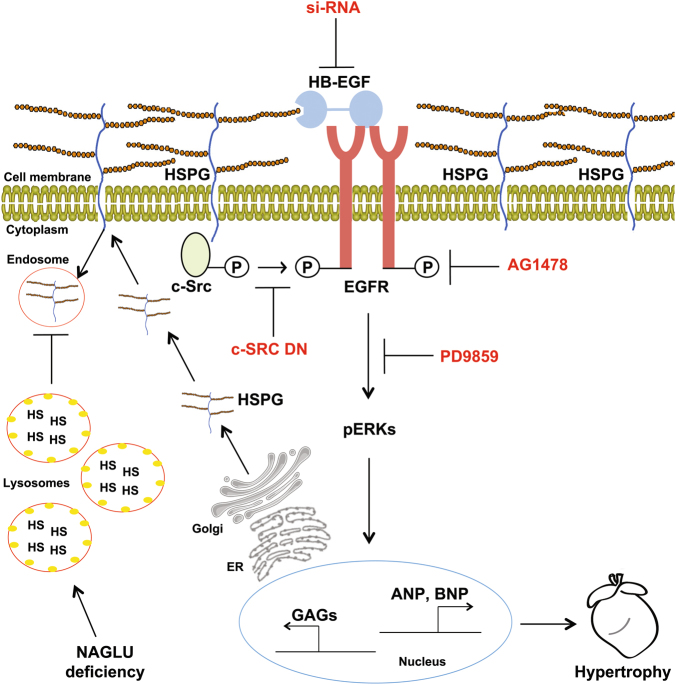
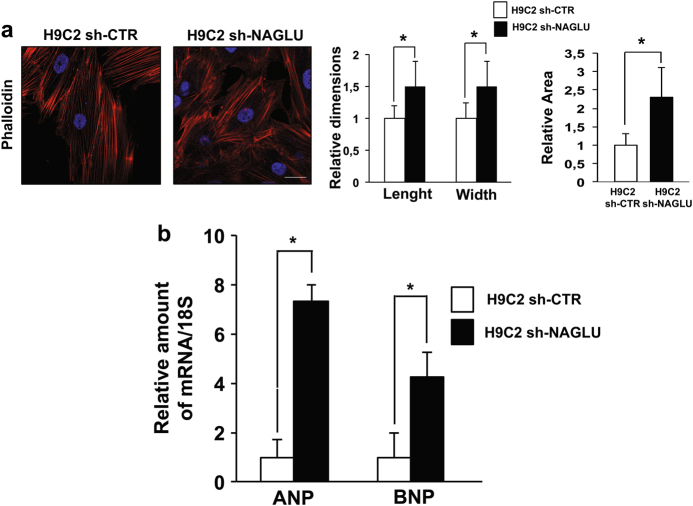
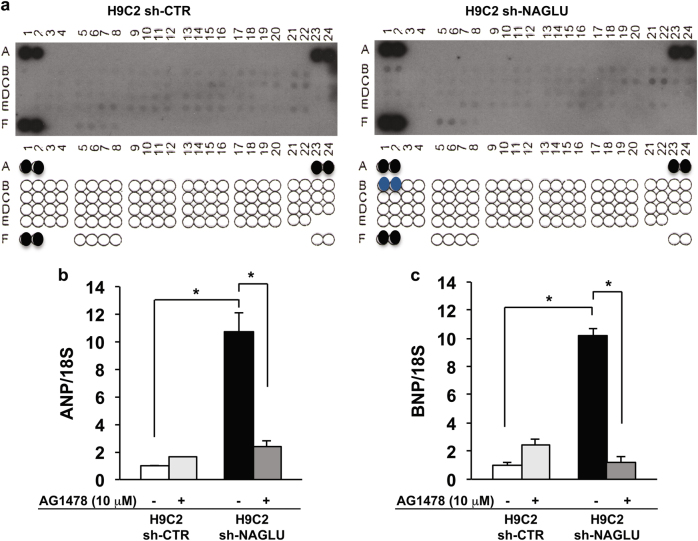
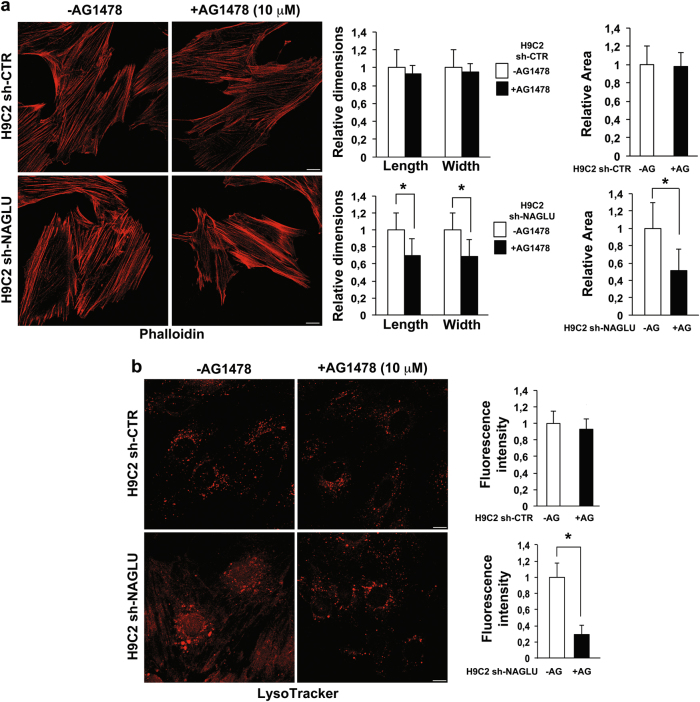
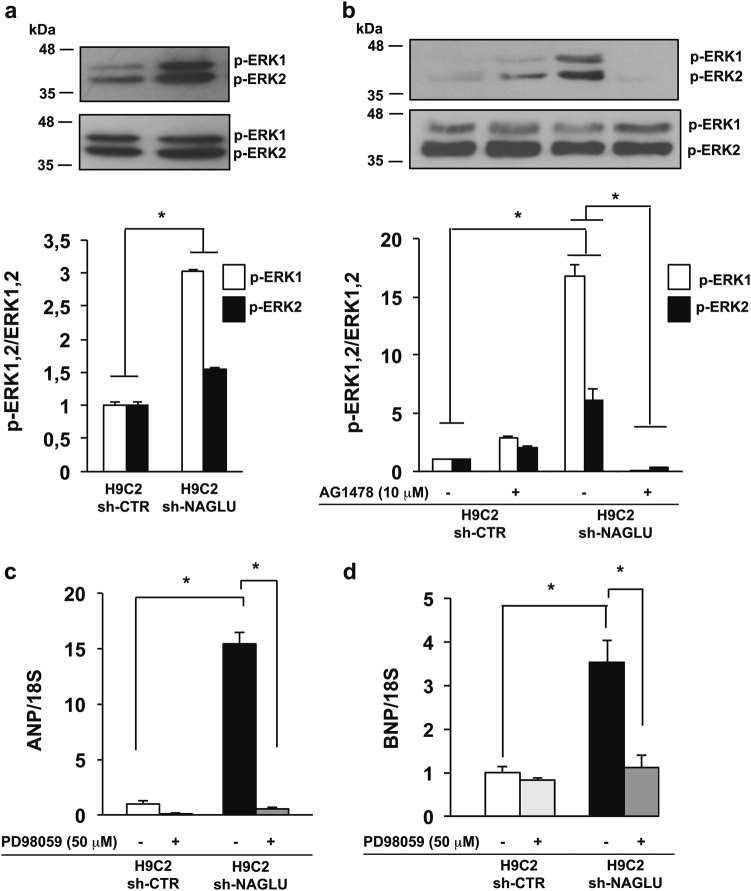
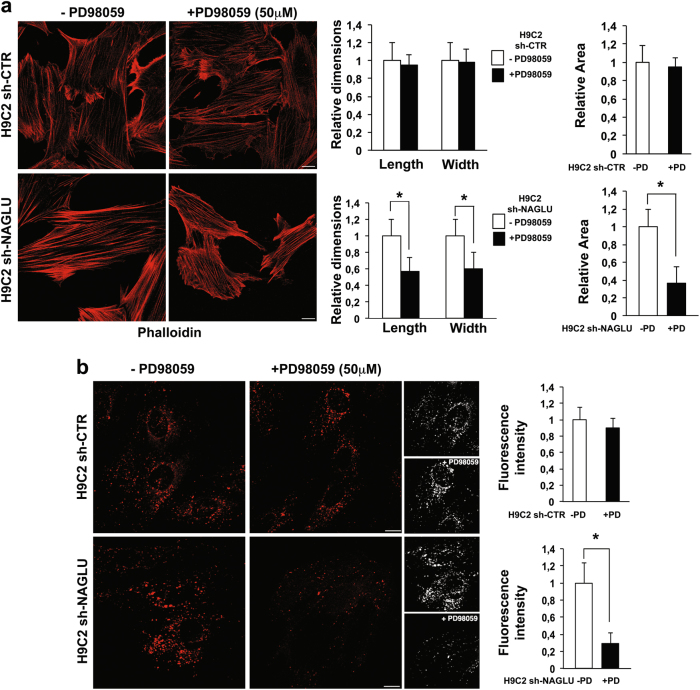
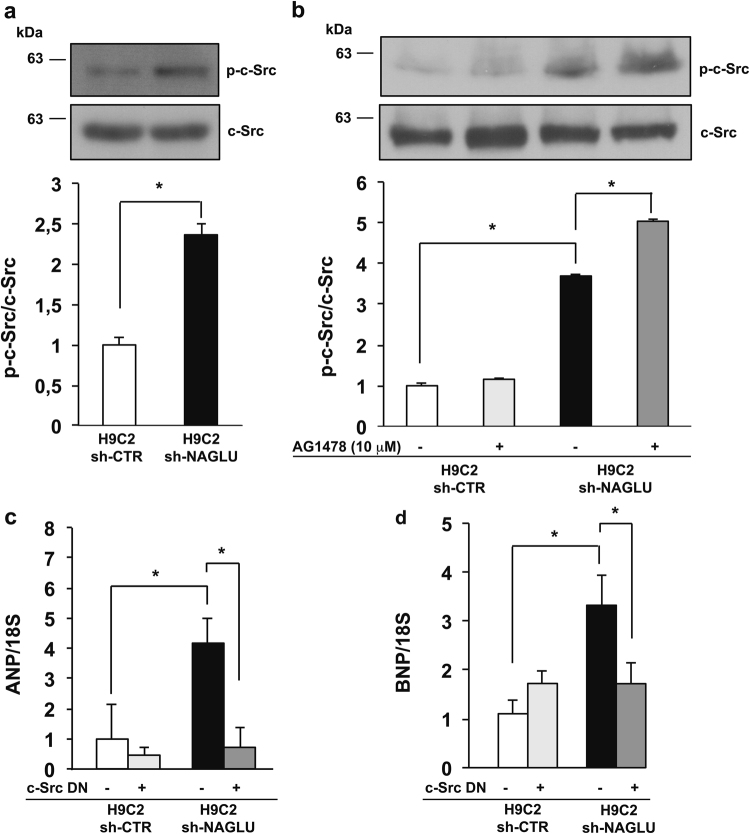
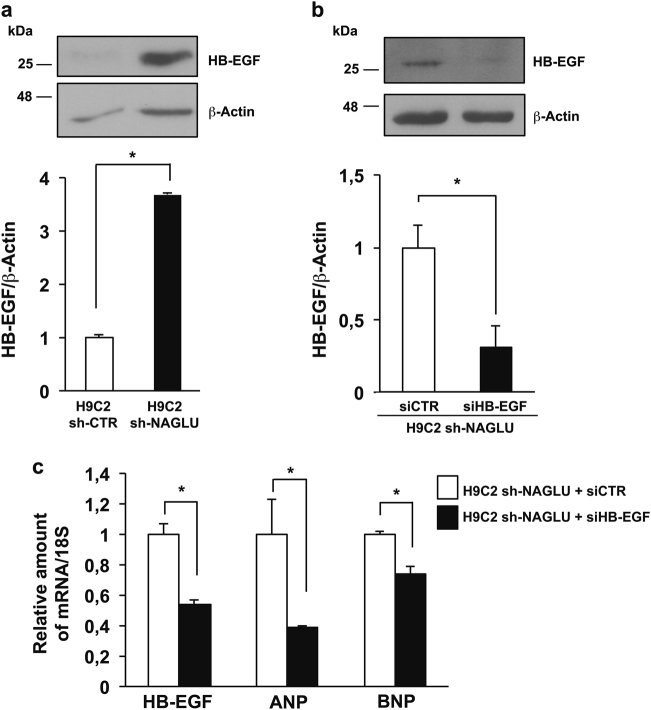
Mucopolysaccharidosis (MPS) IIIB is an inherited lysosomal storage disease caused by the deficiency of the enzyme α-N-acetylglucosaminidase (NAGLU) required for heparan sulfate (HS) degradation. The defective lysosomal clearance of undigested HS results in dysfunction of multiple tissues and organs. We recently demonstrated that the murine model of MPS IIIB develops cardiac disease, valvular abnormalities, and ultimately heart failure. To address the molecular mechanisms governing cardiac dysfunctions in MPS IIIB, we generated a model of the disease by silencing NAGLU gene expression in H9C2 rat cardiomyoblasts. NAGLU-depleted H9C2 exhibited accumulation of abnormal lysosomes and a hypertrophic phenotype. Furthermore, we found the specific activation of the epidermal growth factor receptor (EGFR), and increased phosphorylation levels of extracellular signal-regulated kinases (ERKs) in NAGLU-depleted H9C2. The inhibition of either EGFR or ERKs, using the selective inhibitors AG1478 and PD98059, resulted in the reduction of both lysosomal aberration and hypertrophy in NAGLU-depleted H9C2. We also found increased phosphorylation of c-Src and a reduction of the hypertrophic response in NAGLU-depleted H9C2 transfected with a dominant-negative c-Src. However, c-Src phosphorylation remained unaffected by AG1478 treatment, posing c-Src upstream EGFR activation. Finally, heparin-binding EGF-like growth factor (HB-EGF) protein was found overexpressed in our MPS IIIB cellular model, and its silencing reduced the hypertrophic response. These results indicate that both c-Src and HB-EGF contribute to the hypertrophic phenotype of NAGLU-depleted cardiomyoblasts by synergistically activating EGFR and subsequent signaling, thus suggesting that EGFR pathway inhibition could represent an effective therapeutic approach for MPS IIIB cardiac disease.
黏多糖贮积症(MPS)IIIB 是一种遗传性溶酶体贮积病,由需要降解硫酸乙酰肝素(HS)的酶 α-N-乙酰氨基葡萄糖苷酶(NAGLU)缺乏引起。未消化的 HS 的缺陷性溶酶体清除导致多种组织和器官功能障碍。我们最近证明,MPS IIIB 的小鼠模型会发展出心脏疾病、瓣膜异常,并最终导致心力衰竭。为了解决 MPS IIIB 中心脏功能障碍的分子机制,我们通过沉默 H9C2 大鼠心肌细胞中的 NAGLU 基因表达来建立该疾病的模型。NAGLU 耗尽的 H9C2 表现出异常溶酶体的积累和肥大表型。此外,我们发现 NAGLU 耗尽的 H9C2 中表皮生长因子受体(EGFR)的特异性激活和细胞外信号调节激酶(ERKs)的磷酸化水平增加。使用选择性抑制剂 AG1478 和 PD98059 抑制 EGFR 或 ERKs,可导致 NAGLU 耗尽的 H9C2 中的溶酶体异常和肥大减少。我们还发现,在转染显性失活 c-Src 的 NAGLU 耗尽的 H9C2 中,c-Src 的磷酸化增加,而肥大反应减少。然而,AG1478 处理对 c-Src 的磷酸化没有影响,这表明 c-Src 在上游激活 EGFR。最后,在我们的 MPS IIIB 细胞模型中发现肝素结合表皮生长因子样生长因子(HB-EGF)蛋白过表达,其沉默可减少肥大反应。这些结果表明,c-Src 和 HB-EGF 均通过协同激活 EGFR 和随后的信号传导,导致 NAGLU 耗尽的心肌细胞肥大表型,这表明 EGFR 通路抑制可能是 MPS IIIB 心脏疾病的有效治疗方法。