Department of Chemistry and Alberta Glycomics Centre, University of Alberta, Edmonton, AB T6G 2G2, Canada.
Key Laboratory for NeuroInformation of Ministry of Education, School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu, 610054, China.
Sci Rep. 2018 Jan 19;8(1):1214. doi: 10.1038/s41598-018-19439-2.
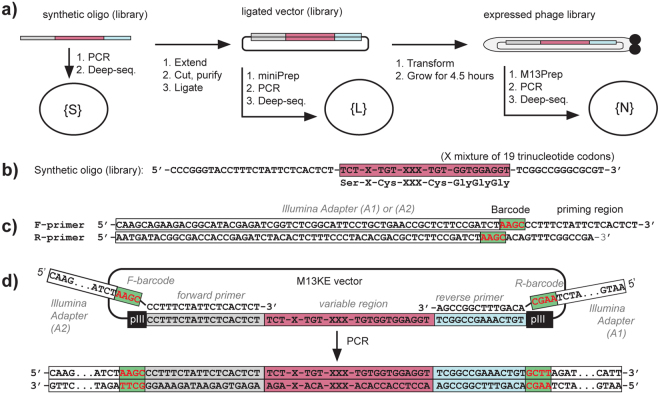
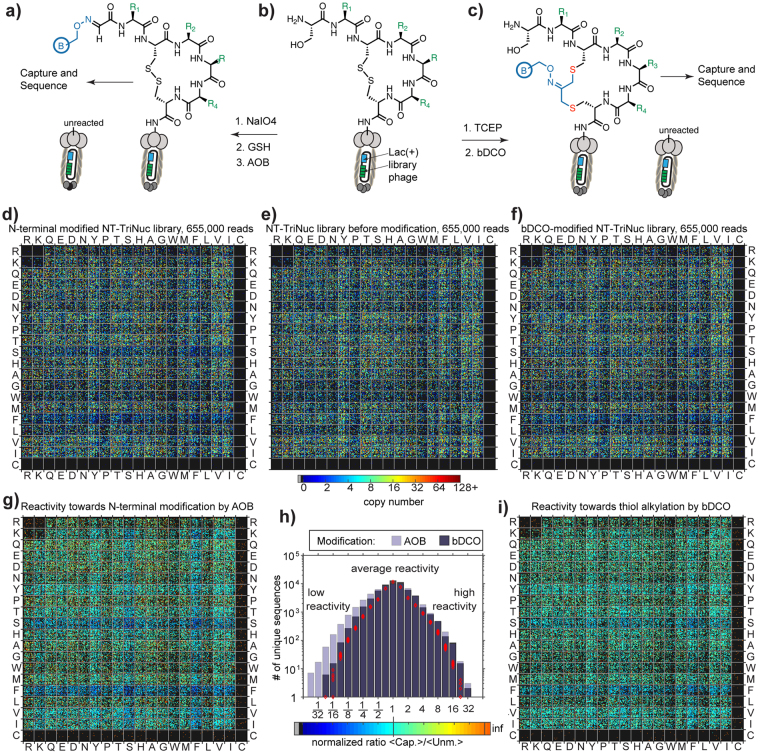
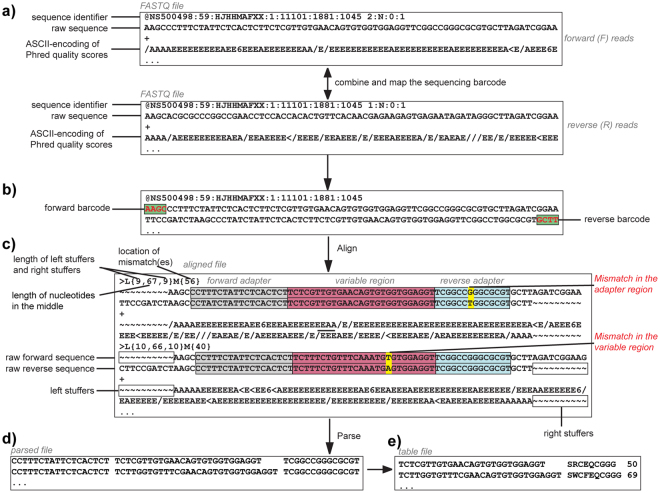
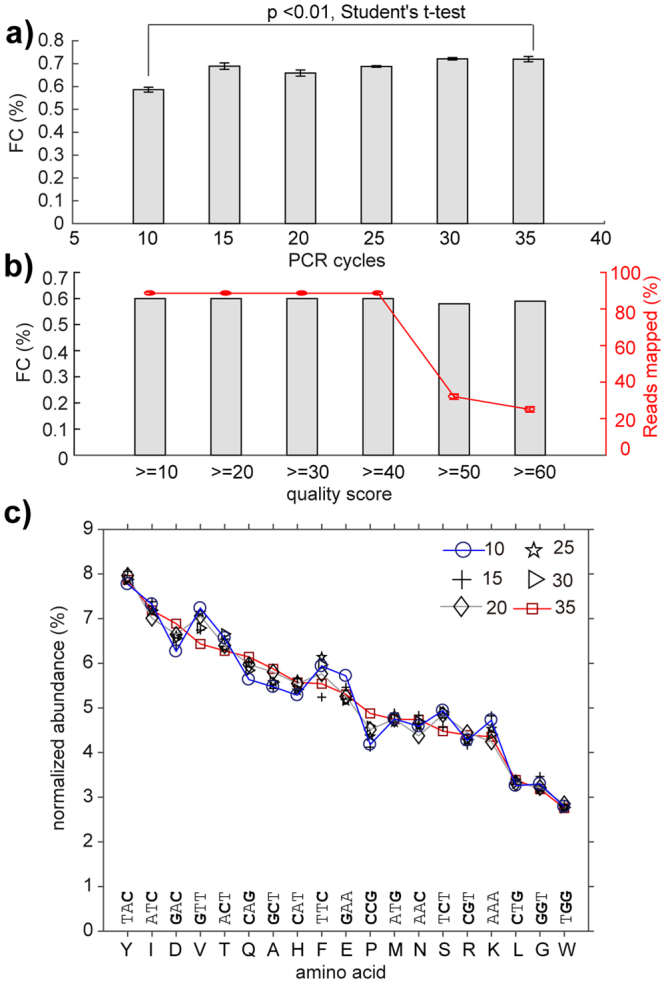
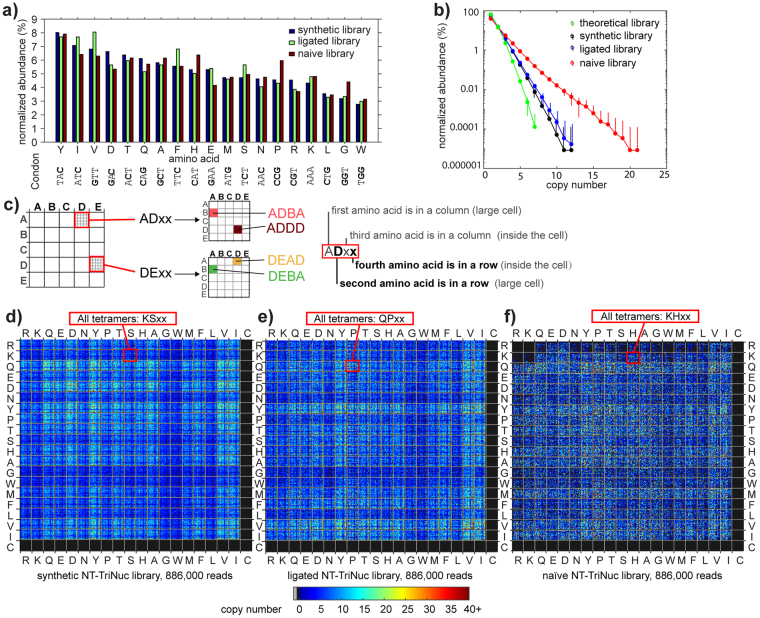
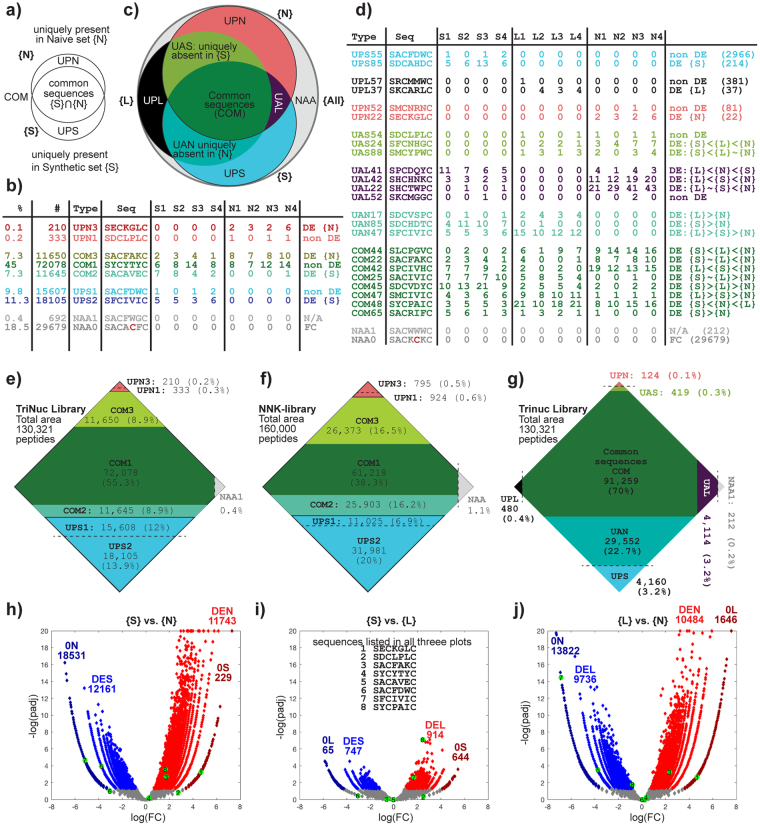
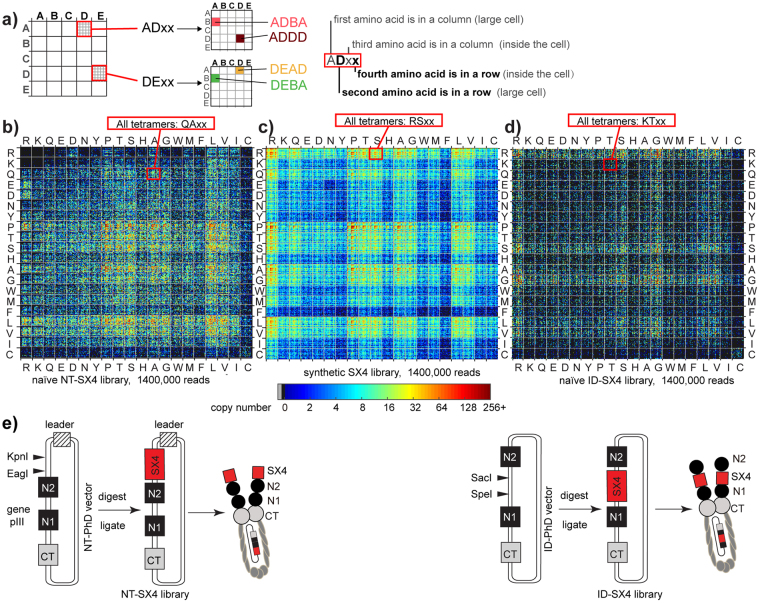
Understanding the composition of a genetically-encoded (GE) library is instrumental to the success of ligand discovery. In this manuscript, we investigate the bias in GE-libraries of linear, macrocyclic and chemically post-translationally modified (cPTM) tetrapeptides displayed on the M13KE platform, which are produced via trinucleotide cassette synthesis (19 codons) and NNK-randomized codon. Differential enrichment of synthetic DNA {S}, ligated vector {L} (extension and ligation of synthetic DNA into the vector), naïve libraries {N} (transformation of the ligated vector into the bacteria followed by expression of the library for 4.5 hours to yield a "naïve" library), and libraries chemically modified by aldehyde ligation and cysteine macrocyclization {M} characterized by paired-end deep sequencing, detected a significant drop in diversity in {L} → {N}, but only a minor compositional difference in {S} → {L} and {N} → {M}. Libraries expressed at the N-terminus of phage protein pIII censored positively charged amino acids Arg and Lys; libraries expressed between pIII domains N1 and N2 overcame Arg/Lys-censorship but introduced new bias towards Gly and Ser. Interrogation of biases arising from cPTM by aldehyde ligation and cysteine macrocyclization unveiled censorship of sequences with Ser/Phe. Analogous analysis can be used to explore library diversity in new display platforms and optimize cPTM of these libraries.
了解基因编码(GE)文库的组成对于配体发现的成功至关重要。在本文中,我们研究了在 M13KE 平台上展示的线性、大环和化学翻译后修饰(cPTM)四肽的 GE 文库中的偏向性,这些文库是通过三核苷酸盒合成(19 个密码子)和 NNK 随机化密码子产生的。通过配对末端深度测序对合成 DNA{S}、连接载体{L}(将合成 DNA 连接到载体上的延伸和连接)、原始文库{N}(连接载体转化为细菌,然后表达文库 4.5 小时以产生“原始”文库)和通过醛连接和半胱氨酸环化化学修饰的文库{M}进行了差异富集,检测到{L}→{N}中的多样性显著下降,但{S}→{L}和{N}→{M}中的组成差异很小。在噬菌体蛋白 pIII 的 N 末端表达的文库中,正电荷氨基酸 Arg 和 Lys 受到了审查;在 pIII 结构域 N1 和 N2 之间表达的文库克服了 Arg/Lys 审查,但引入了对 Gly 和 Ser 的新偏向性。通过醛连接和半胱氨酸环化进行 cPTM 引起的偏向性的探究揭示了 Ser/Phe 序列的审查。类似的分析可用于探索新展示平台中的文库多样性,并优化这些文库的 cPTM。