Department of Clinical Physiology and Nuclear Medicine & Cluster for Molecular Imaging, Copenhagen University Hospital-Rigshospitalet & Department of Biomedical Sciences, University of Copenhagen, 2200 Copenhagen, Denmark.
Nano-Science Center, Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark.
Viruses. 2022 Oct 29;14(11):2402. doi: 10.3390/v14112402.
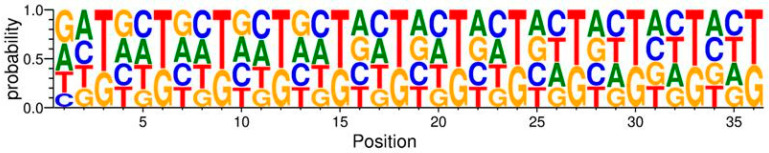
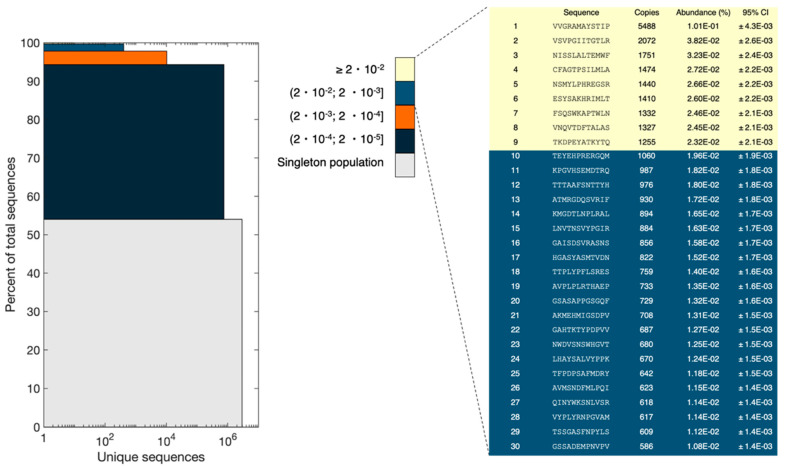
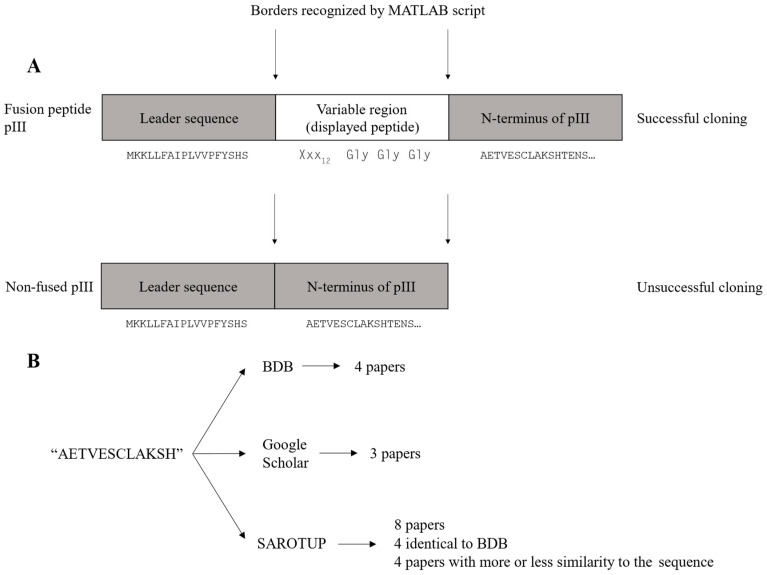
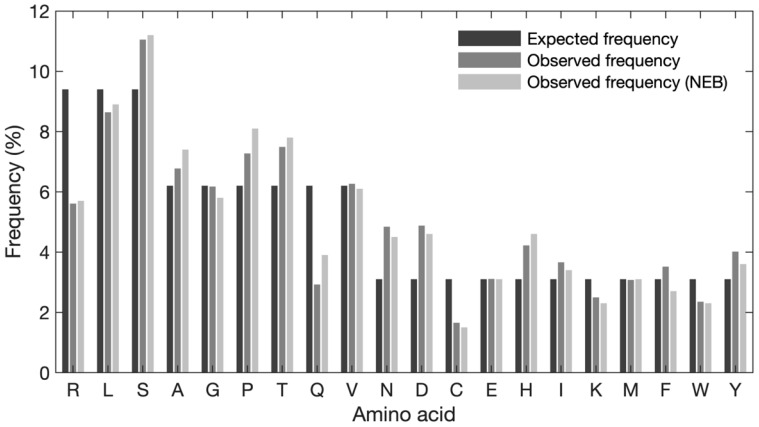
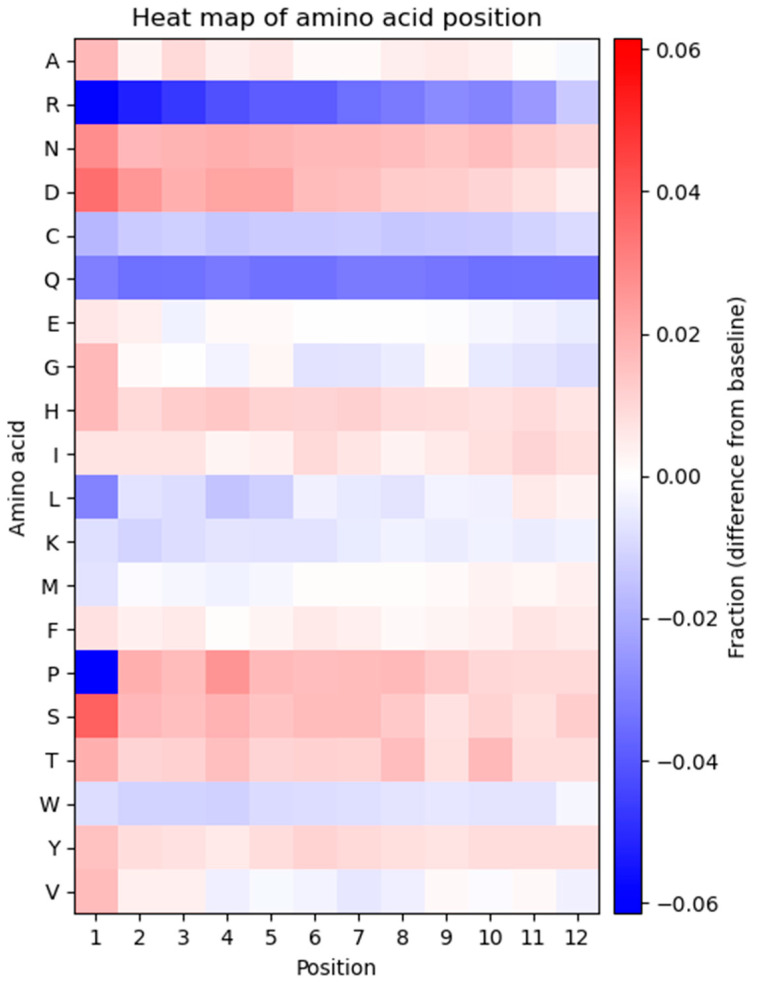
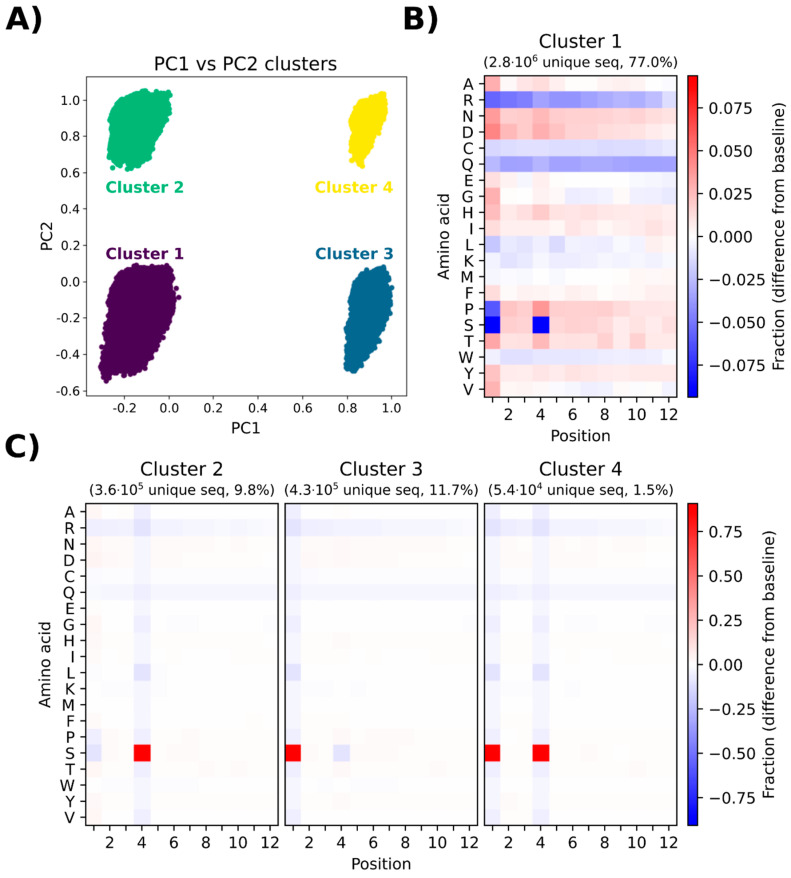
The principal presumption of phage display biopanning is that the naïve library contains an unbiased repertoire of peptides, and thus, the enriched variants derive from the affinity selection of an entirely random peptide pool. In the current study, we utilized deep sequencing to characterize the widely used Ph.DTM-12 phage display peptide library (New England Biolabs). The next-generation sequencing (NGS) data indicated the presence of stop codons and a high abundance of wild-type clones in the naïve library, which collectively result in a reduced effective size of the library. The analysis of the DNA sequence logo and global and position-specific frequency of amino acids demonstrated significant bias in the nucleotide and amino acid composition of the library inserts. Principal component analysis (PCA) uncovered the existence of four distinct clusters in the naïve library and the investigation of peptide frequency distribution revealed a broad range of unequal abundances for peptides. Taken together, our data provide strong evidence for the notion that the naïve library represents substantial departures from randomness at the nucleotide, amino acid, and peptide levels, though not undergoing any selective pressure for target binding. This non-uniform sequence representation arises from both the M13 phage biology and technical errors of the library construction. Our findings highlight the paramount importance of the qualitative assessment of the naïve phage display libraries prior to biopanning.
噬菌体展示生物淘选的主要假设是,原始文库包含了一个无偏倚的肽库,因此,富集的变体来源于对完全随机肽库的亲和选择。在本研究中,我们利用深度测序来描述广泛使用的 Ph.DTM-12 噬菌体展示肽文库(New England Biolabs)。下一代测序(NGS)数据表明,原始文库中存在终止密码子和大量野生型克隆,这共同导致文库的有效大小减小。对 DNA 序列标志和文库插入物中核苷酸和氨基酸的全局和位置特异性频率的分析表明,文库插入物的核苷酸和氨基酸组成存在显著的偏倚。主成分分析(PCA)揭示了原始文库中存在四个不同的簇,对肽频率分布的研究表明,肽的丰度存在广泛的不均匀性。总之,我们的数据为以下观点提供了强有力的证据,即原始文库在核苷酸、氨基酸和肽水平上都存在显著偏离随机性的情况,尽管没有针对靶标结合进行任何选择压力。这种非均匀的序列表示既来自 M13 噬菌体生物学,也来自文库构建的技术误差。我们的研究结果强调了在进行生物淘选之前,对原始噬菌体展示文库进行定性评估的至关重要性。