Department of Health Technology and Informatics, The Hong Kong Polytechnic University, Hong Kong, Hong Kong.
Department of Microbiology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, Hong Kong.
Front Cell Infect Microbiol. 2018 Jan 12;7:539. doi: 10.3389/fcimb.2017.00539. eCollection 2017.
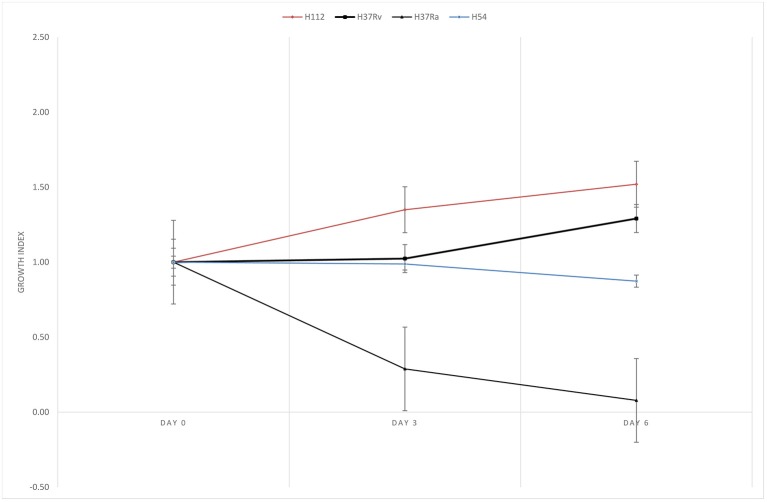
Development of improved therapeutics against tuberculosis (TB) is hindered by an inadequate understanding of the relationship between disease severity and genetic diversity of its causative agent, . We previously isolated a hypervirulent strain H112 from an HIV-negative patient with an aggressive disease progression from pulmonary TB to tuberculous meningitis-the most severe manifestation of tuberculosis. Human macrophage challenge experiment demonstrated that the strain H112 exhibited significantly better intracellular survivability and induced lower level of TNF-α than the reference virulent strain and other 123 clinical isolates. The present study aimed to identify the potential genetic determinants of mycobacterial virulence that were common to strain H112 and hypervirulent strains of the same phylogenetic clade isolated in other global regions. A low-virulent strain H54 which belonged to the same phylogenetic lineage (L2) as strain H112 was selected from a collection of 115 clinical isolates. Both H112 and H54 were whole-genome-sequenced using PacBio sequencing technology. A comparative genomics approach was adopted to identify mutations present in strain H112 but absent in strain H54. Subsequently, an extensive phylogenetic analysis was conducted by including all publically available genomes. Single-nucleotide-polymorphisms (SNPs) and structural variations (SVs) common to hypervirulent strains in the global collection of genomes were considered as potential genetic determinants of hypervirulence. Sequencing data revealed that both H112 and H54 were identified as members of the same sub-lineage L2.2.1. After excluding the lineage-related mutations shared between H112 and H54, we analyzed the phylogenetic relatedness of H112 with global collection of genomes ( = 4,338), and identified a novel phylogenetic clade in which four hypervirulent strains isolated from geographically diverse regions were clustered together. All hypervirulent strains in the clade shared 12 SNPs and 5 SVs with H112, including those affecting key virulence-associated loci, notably, a deleterious SNP ( p. D150E) within operon and an intergenic deletion (854259_ 854261delCC) in close-proximity to . The present study identified common genetic factors in a novel phylogenetic clade of hypervirulent . The causative role of these mutations in mycobacterial virulence should be validated in future study.
针对结核病(TB)的改良疗法的发展受到对其病原体疾病严重程度和遗传多样性之间关系的理解不足的阻碍。我们之前从一名患有 HIV 阴性、疾病进展迅速的肺结核患者中分离出了一株高度毒力的菌株 H112,该患者从肺结核发展为结核性脑膜炎——这是结核病最严重的表现形式。人类巨噬细胞挑战实验表明,与参考毒力菌株 H37Rv 和其他 123 株临床分离株相比,该菌株 H112 表现出更好的细胞内存活率和更低水平的 TNF-α。本研究旨在确定与 H112 和来自其他全球地区同一进化枝的高度毒力菌株共同存在的分枝杆菌毒力的潜在遗传决定因素。一株低毒力的菌株 H54 来自一个包含 115 株临床分离株的收藏库,它与菌株 H112 属于同一进化谱系(L2)。使用 PacBio 测序技术对 H112 和 H54 进行全基因组测序。采用比较基因组学方法来鉴定存在于菌株 H112 中而不存在于菌株 H54 中的突变。随后,通过包括所有公开的基因组,进行了广泛的系统发育分析。将在全球基因组收藏中高度毒力菌株共同存在的单核苷酸多态性(SNP)和结构变异(SV)视为高度毒力的潜在遗传决定因素。测序数据显示,H112 和 H54 均被鉴定为同一亚谱系 L2.2.1 的成员。在排除 H112 和 H54 之间共享的谱系相关突变后,我们分析了 H112 与全球基因组收藏(n=4338)的系统发育关系,并在一个新的进化枝中鉴定了四个来自地理上不同地区的高度毒力菌株聚集在一起。该进化枝中的所有高度毒力菌株与 H112 共有 12 个 SNP 和 5 个 SV,包括影响关键毒力相关基因座的 SNP,特别是 operon 内的有害 SNP(p.D150E)和紧密临近处的基因间缺失(854259_854261delCC)。本研究在一个新的高度毒力分枝杆菌的进化枝中确定了共同的遗传因素。这些突变在分枝杆菌毒力中的因果作用应在未来的研究中得到验证。