Schaefer Christiane, Mallela Nikhil, Seggewiß Jochen, Lechtape Birgit, Omran Heymut, Dirksen Uta, Korsching Eberhard, Potratz Jenny
Pediatric Hematology and Oncology, University Hospital Münster, Münster, Germany.
Institute of Bioinformatics, Westfälische Wilhelms-Universität Münster, Münster, Germany.
PLoS One. 2018 Jan 31;13(1):e0191570. doi: 10.1371/journal.pone.0191570. eCollection 2018.
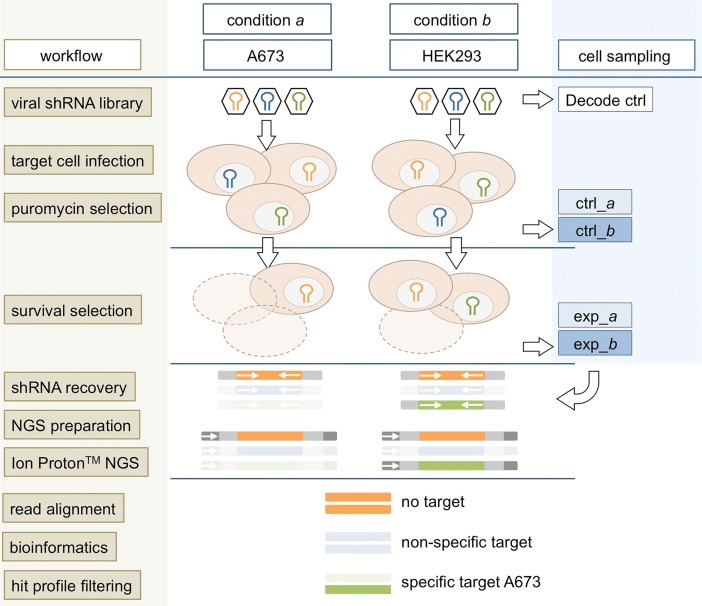
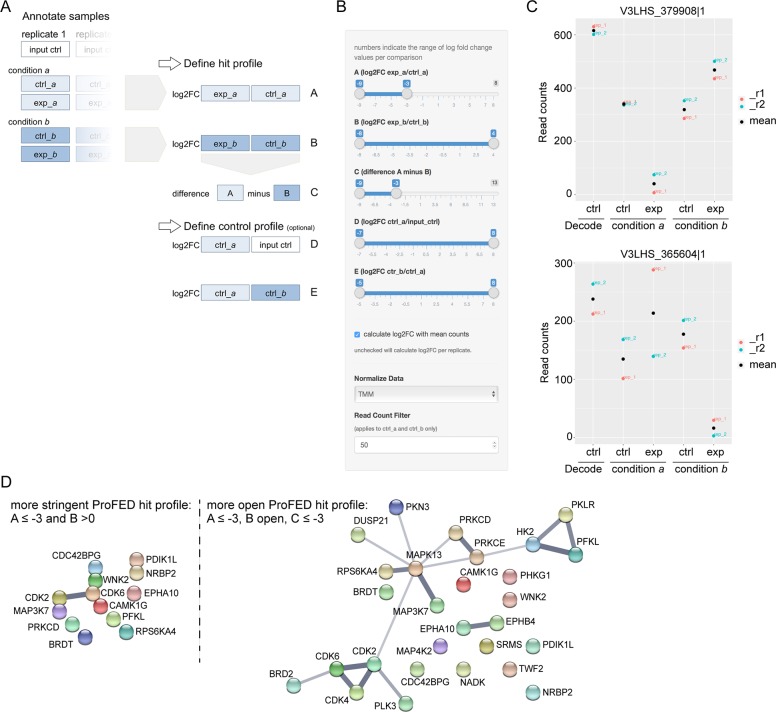
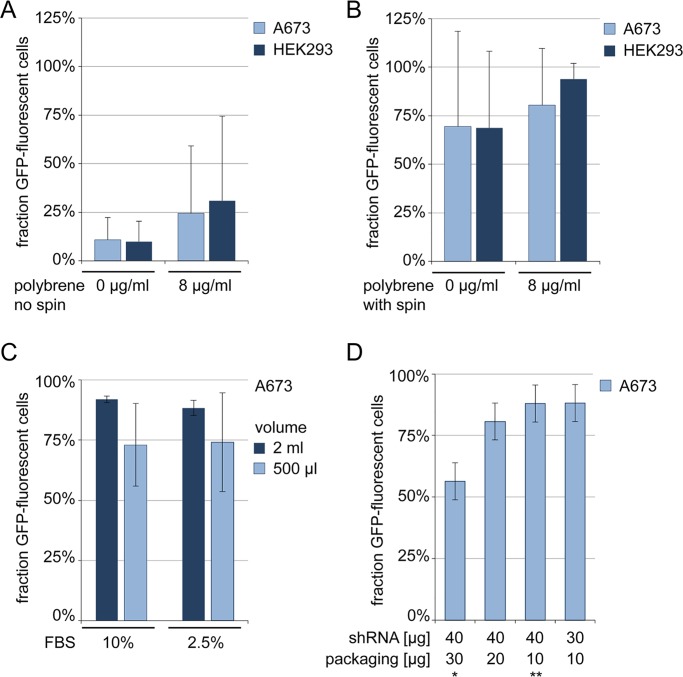
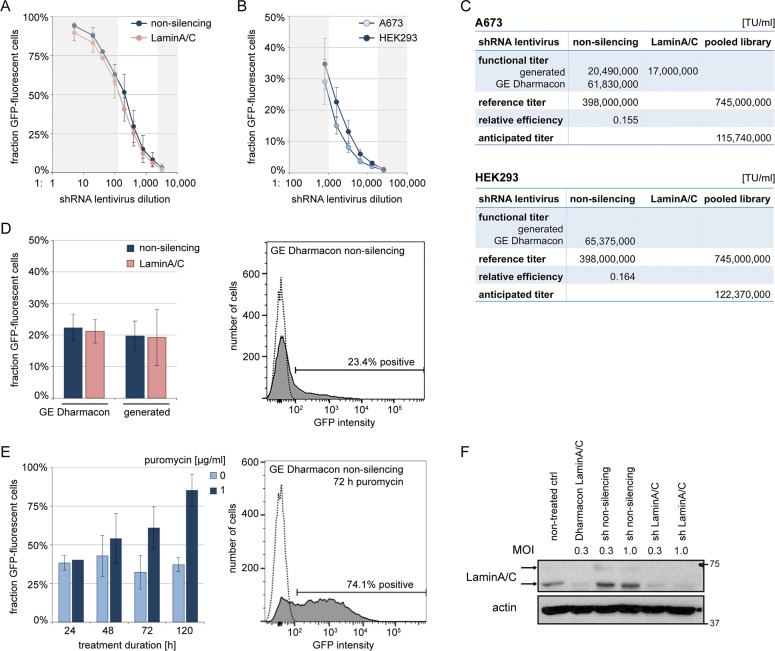
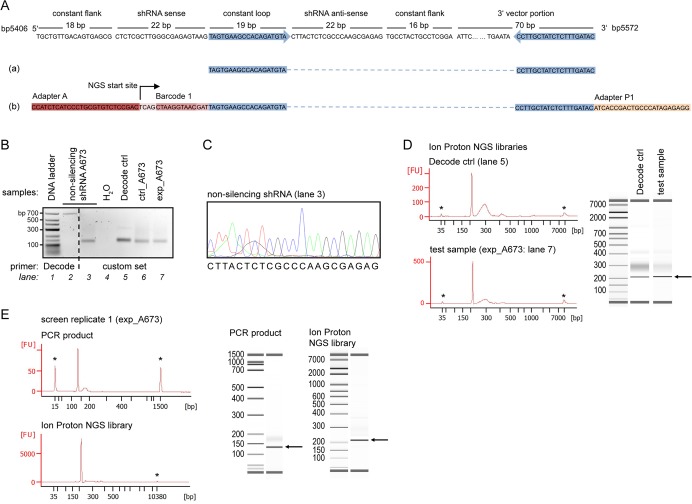
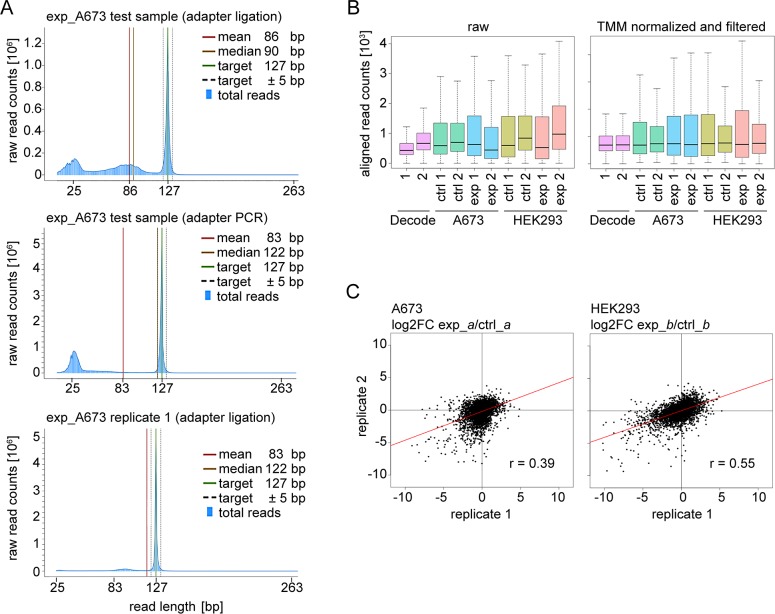
In the search for novel therapeutic targets, RNA interference screening has become a valuable tool. High-throughput technologies are now broadly accessible but their assay development from baseline remains resource-intensive and challenging. Focusing on this assay development process, we here describe a target discovery screen using pooled shRNA libraries and next-generation sequencing (NGS) deconvolution in a cell line model of Ewing sarcoma. In a strategy designed for comparative and synthetic lethal studies, we screened for targets specific to the A673 Ewing sarcoma cell line. Methods, results and pitfalls are described for the entire multi-step screening procedure, from lentiviral shRNA delivery to bioinformatics analysis, illustrating a complete model workflow. We demonstrate that successful studies are feasible from the first assay performance and independent of specialized screening units. Furthermore, we show that a resource-saving screen depth of 100-fold average shRNA representation can suffice to generate reproducible target hits despite heterogeneity in the derived datasets. Because statistical analysis methods are debatable for such datasets, we created ProFED, an analysis package designed to facilitate descriptive data analysis and hit calling using an aim-oriented profile filtering approach. In its versatile design, this open-source online tool provides fast and easy analysis of shRNA and other count-based datasets to complement other analytical algorithms.
在寻找新型治疗靶点的过程中,RNA干扰筛选已成为一种有价值的工具。高通量技术如今已广泛可用,但从基线开始进行实验开发仍然资源密集且具有挑战性。针对这一实验开发过程,我们在此描述了一种在尤因肉瘤细胞系模型中使用混合shRNA文库和下一代测序(NGS)解卷积的靶点发现筛选方法。在一项为比较性和合成致死性研究设计的策略中,我们筛选了A673尤因肉瘤细胞系特有的靶点。本文描述了从慢病毒shRNA递送至生物信息学分析的整个多步骤筛选程序的方法、结果和陷阱,展示了一个完整的模型工作流程。我们证明,从首次实验性能来看,成功的研究是可行的,且无需专门的筛选单位。此外,我们表明,尽管衍生数据集中存在异质性,但平均shRNA代表性为100倍的资源节约型筛选深度足以产生可重复的靶点命中结果。由于此类数据集的统计分析方法存在争议,我们创建了ProFED,这是一个分析包,旨在使用面向目标的轮廓过滤方法促进描述性数据分析和命中调用。在其通用设计中,这个开源在线工具提供了对shRNA和其他基于计数的数据集的快速简便分析,以补充其他分析算法。