Yuan Yongxian, Xu Huaiqian, Leung Ross Ka-Kit
BGI-tech, BGI-Shenzhen, Shenzhen, 518083, Guangdong, China.
BGI-tech, BGI-Wuhan, Wuhan, 430075, Hubei, China.
BMC Genomics. 2016 May 26;17:403. doi: 10.1186/s12864-016-2745-8.
Previous studies compared running cost, time and other performance measures of popular sequencing platforms. However, comprehensive assessment of library construction and analysis protocols for Proton sequencing platform remains unexplored. Unlike Illumina sequencing platforms, Proton reads are heterogeneous in length and quality. When sequencing data from different platforms are combined, this can result in reads with various read length. Whether the performance of the commonly used software for handling such kind of data is satisfactory is unknown.
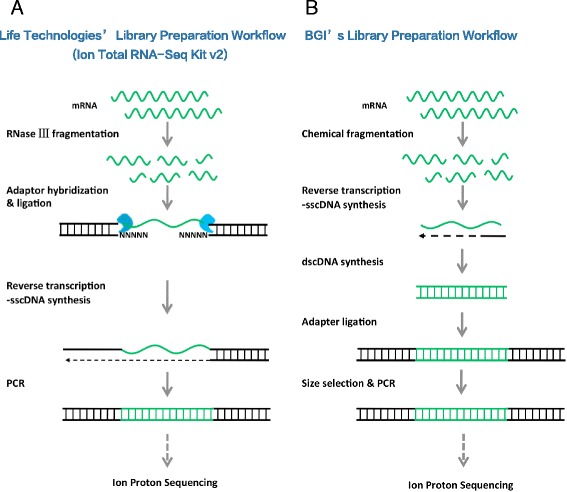
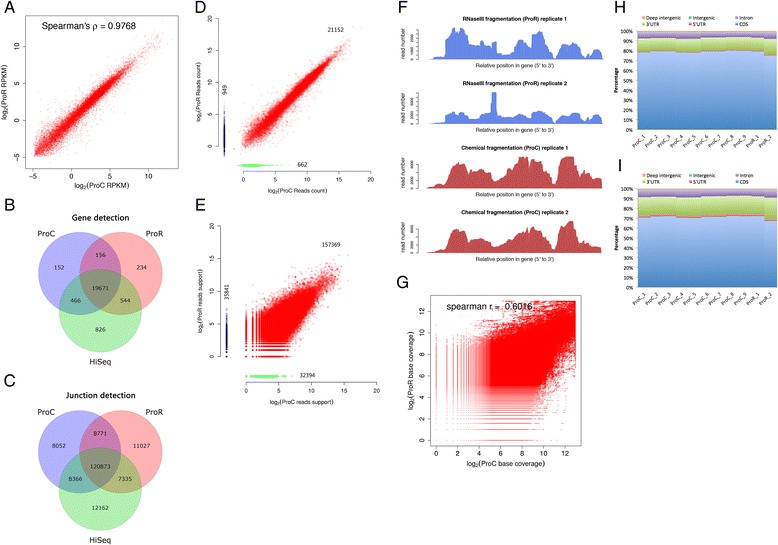
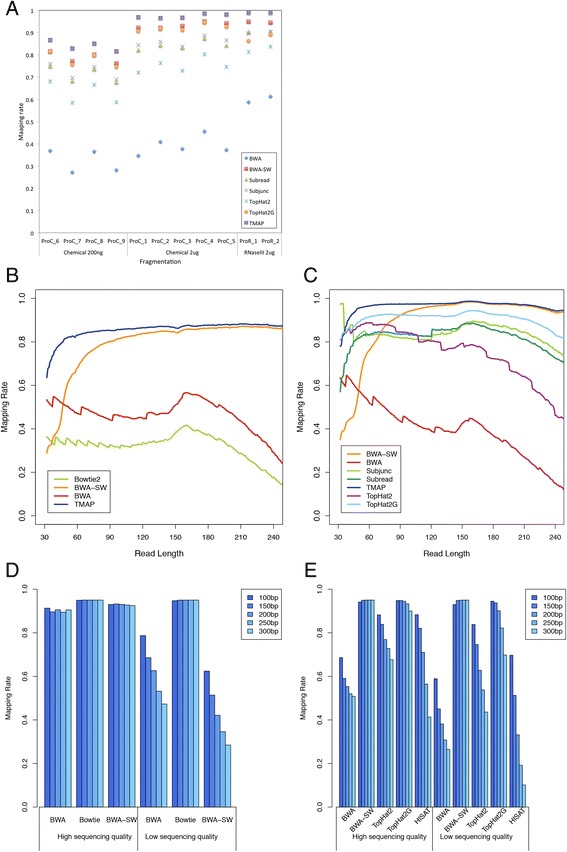
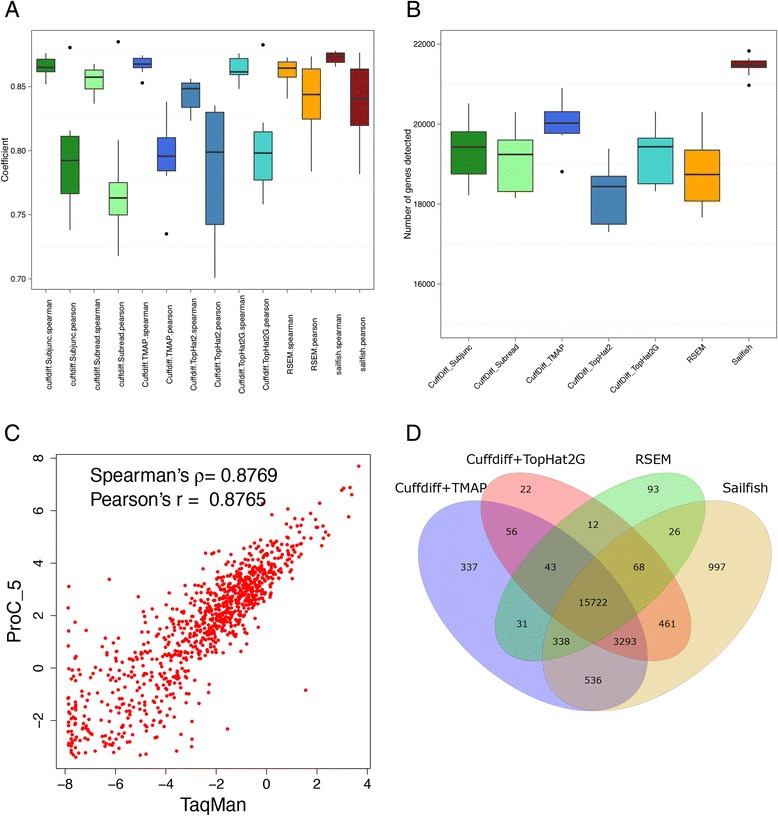
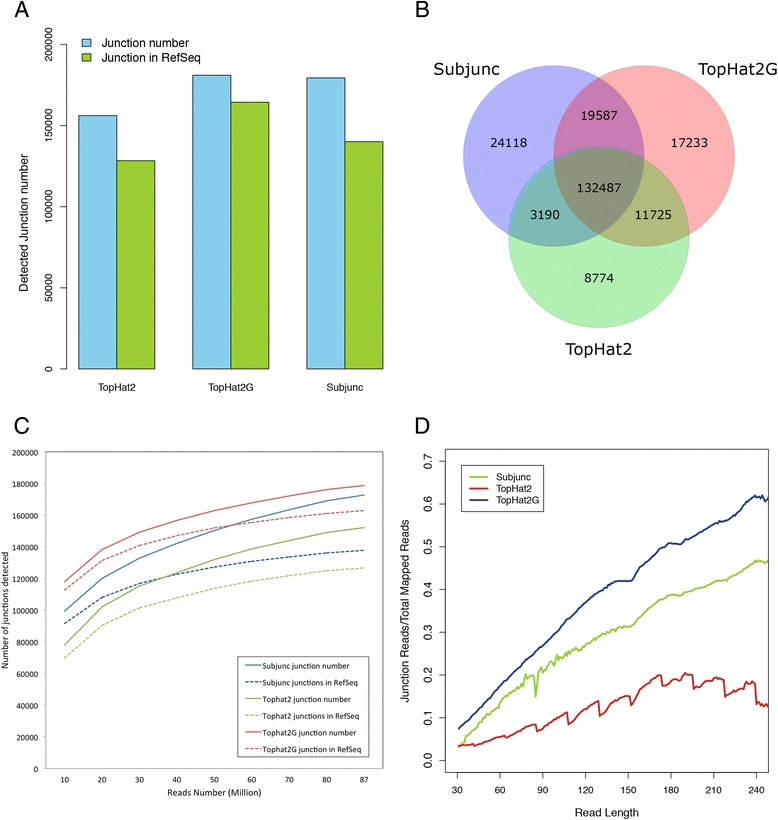
By using universal human reference RNA as the initial material, RNaseIII and chemical fragmentation methods in library construction showed similar result in gene and junction discovery number and expression level estimated accuracy. In contrast, sequencing quality, read length and the choice of software affected mapping rate to a much larger extent. Unspliced aligner TMAP attained the highest mapping rate (97.27 % to genome, 86.46 % to transcriptome), though 47.83 % of mapped reads were clipped. Long reads could paradoxically reduce mapping in junctions. With reference annotation guide, the mapping rate of TopHat2 significantly increased from 75.79 to 92.09 %, especially for long (>150 bp) reads. Sailfish, a k-mer based gene expression quantifier attained highly consistent results with that of TaqMan array and highest sensitivity.
We provided for the first time, the reference statistics of library preparation methods, gene detection and quantification and junction discovery for RNA-Seq by the Ion Proton platform. Chemical fragmentation performed equally well with the enzyme-based one. The optimal Ion Proton sequencing options and analysis software have been evaluated.
以往的研究比较了流行测序平台的运行成本、时间及其他性能指标。然而,对Proton测序平台的文库构建和分析方案的全面评估仍未开展。与Illumina测序平台不同,Proton测序读段的长度和质量存在异质性。当合并来自不同平台的测序数据时,这可能导致产生具有各种读长的读段。处理这类数据的常用软件的性能是否令人满意尚不清楚。
以通用人类参考RNA作为起始材料,文库构建中RNaseIII和化学片段化方法在基因和接头发现数量以及表达水平估计准确性方面显示出相似的结果。相比之下,测序质量、读长和软件选择对比对率的影响要大得多。未剪接比对软件TMAP获得了最高的比对率(对基因组为97.27%,对转录组为86.46%),不过47.83%的比对读段被剪切。长读段反而会降低接头处的比对率。借助参考注释指南,TopHat2的比对率从7(此处原文有误,应为75.79)显著提高到92.09%,尤其是对于长度大于150bp的长读段。基于k-mer的基因表达定量软件Sailfish与TaqMan芯片获得了高度一致的结果,且灵敏度最高。
我们首次提供了Ion Proton平台RNA-Seq文库制备方法、基因检测与定量以及接头发现的参考统计数据。化学片段化与基于酶的方法表现相当。我们评估了Ion Proton测序的最佳选择和分析软件。