Mallinckrodt Institute of Radiology, Washington University, St Louis, MO, USA; Knight Alzheimer's Disease Research Center, Washington University, St Louis, MO, USA; Department of Psychological & Brain Sciences, Washington University, St Louis, MO, USA.
Mallinckrodt Institute of Radiology, Washington University, St Louis, MO, USA; Division of Biology and Biomedical Sciences, Washington University, St Louis, MO, USA.
Lancet Neurol. 2018 Mar;17(3):241-250. doi: 10.1016/S1474-4422(18)30028-0. Epub 2018 Feb 1.
Models of Alzheimer's disease propose a sequence of amyloid β (Aβ) accumulation, hypometabolism, and structural decline that precedes the onset of clinical dementia. These pathological features evolve both temporally and spatially in the brain. In this study, we aimed to characterise where in the brain and when in the course of the disease neuroimaging biomarkers become abnormal.
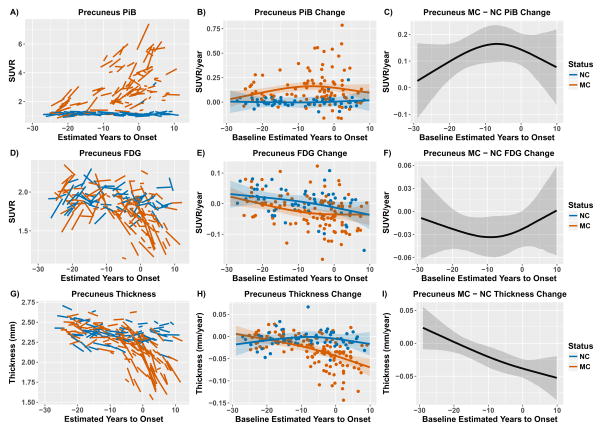
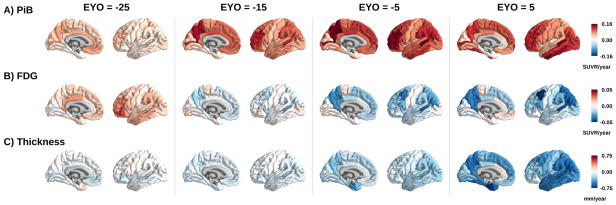
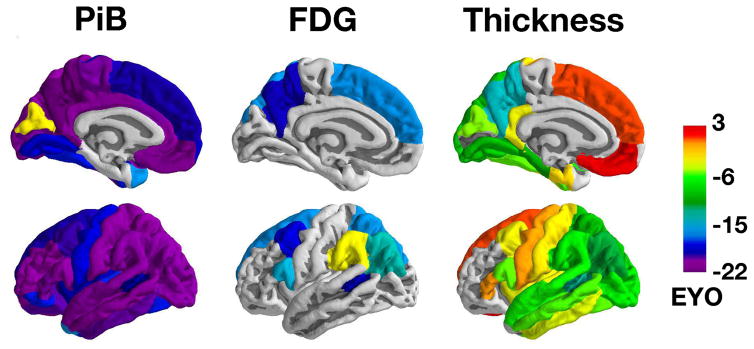
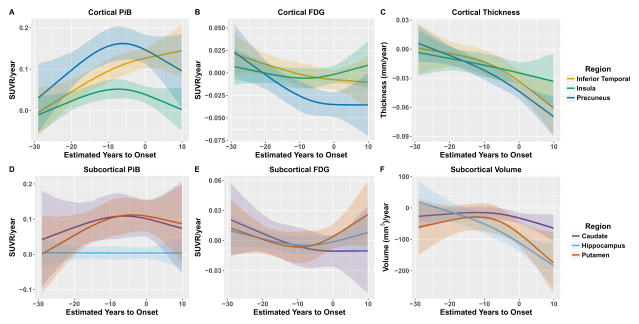
Between Jan 1, 2009, and Dec 31, 2015, we analysed data from mutation non-carriers, asymptomatic carriers, and symptomatic carriers from families carrying gene mutations in presenilin 1 (PSEN1), presenilin 2 (PSEN2), or amyloid precursor protein (APP) enrolled in the Dominantly Inherited Alzheimer's Network. We analysed C-Pittsburgh Compound B (C-PiB) PET, F-Fluorodeoxyglucose (F-FDG) PET, and structural MRI data using regions of interest to assess change throughout the brain. We estimated rates of biomarker change as a function of estimated years to symptom onset at baseline using linear mixed-effects models and determined the earliest point at which biomarker trajectories differed between mutation carriers and non-carriers. This study is registered at ClinicalTrials.gov (number NCT00869817) FINDINGS: C-PiB PET was available for 346 individuals (162 with longitudinal imaging), F-FDG PET was available for 352 individuals (175 with longitudinal imaging), and MRI data were available for 377 individuals (201 with longitudinal imaging). We found a sequence to pathological changes, with rates of Aβ deposition in mutation carriers being significantly different from those in non-carriers first (across regions that showed a significant difference, at a mean of 18·9 years [SD 3·3] before expected onset), followed by hypometabolism (14·1 years [5·1] before expected onset), and lastly structural decline (4·7 years [4·2] before expected onset). This biomarker ordering was preserved in most, but not all, regions. The temporal emergence within a biomarker varied across the brain, with the precuneus being the first cortical region for each method to show divergence between groups (22·2 years before expected onset for Aβ accumulation, 18·8 years before expected onset for hypometabolism, and 13·0 years before expected onset for cortical thinning).
Mutation carriers had elevations in Aβ deposition, reduced glucose metabolism, and cortical thinning compared with non-carriers which preceded the expected onset of dementia. Accrual of these pathologies varied throughout the brain, suggesting differential regional and temporal vulnerabilities to Aβ, metabolic decline, and structural atrophy, which should be taken into account when using biomarkers in a clinical setting as well as designing and evaluating clinical trials.
US National Institutes of Health, the German Center for Neurodegenerative Diseases, and the Medical Research Council Dementias Platform UK.
阿尔茨海默病模型提出了淀粉样β(Aβ)积累、低代谢和结构下降的序列,这些病理特征在大脑中既具有时间性又具有空间性。在这项研究中,我们旨在描述神经影像学生物标志物在疾病过程中何时何地变得异常。
在 2009 年 1 月 1 日至 2015 年 12 月 31 日期间,我们分析了携带早老素 1(PSEN1)、早老素 2(PSEN2)或淀粉样前体蛋白(APP)基因突变的非突变携带者、无症状携带者和有症状携带者的家族成员的数据,这些成员参与了显性遗传性阿尔茨海默病网络。我们使用感兴趣区域分析 C-Pittsburgh 复合 B(C-PiB)PET、F-氟脱氧葡萄糖(F-FDG)PET 和结构 MRI 数据,以评估整个大脑的变化。我们使用线性混合效应模型,根据基线时预计发病时间,估计生物标志物变化的速度,并确定生物标志物轨迹在突变携带者和非携带者之间最早不同的时间点。这项研究在 ClinicalTrials.gov 注册(编号 NCT00869817)。
C-PiB PET 可用于 346 人(162 人进行纵向成像),F-FDG PET 可用于 352 人(175 人进行纵向成像),MRI 数据可用于 377 人(201 人进行纵向成像)。我们发现了一个病理变化的序列,突变携带者的 Aβ沉积率与非携带者的沉积率有显著差异(在显示显著差异的区域,平均在预期发病前 18.9 年[3.3 年]),其次是低代谢(14.1 年[5.1 年]),最后是结构下降(4.7 年[4.2 年])。这种生物标志物的排序在大多数但不是所有区域都得到了保留。在大脑内,每种方法的生物标志物的出现时间都有所不同,距预计发病时间最近的是扣带回前部(Aβ 积累的预计发病时间为 22.2 年,低代谢的预计发病时间为 18.8 年,皮质变薄的预计发病时间为 13.0 年)。
与非携带者相比,突变携带者的 Aβ 沉积、葡萄糖代谢减少和皮质变薄升高,这些变化都发生在痴呆预期发病之前。这些病理变化在整个大脑中积累,这表明 Aβ、代谢下降和结构萎缩在不同的区域和时间存在易感性,在临床环境中使用生物标志物以及设计和评估临床试验时,都应考虑到这一点。
美国国立卫生研究院、德国神经退行性疾病中心和英国医学研究理事会痴呆症平台。