Department of Pharmacology (the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Harbin, 150081, China.
Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (CAMS) and Comparative Medicine Centre, Peking Union Medical Collage (PUMC), Beijing, China.
Cell Death Dis. 2018 Feb 7;9(2):171. doi: 10.1038/s41419-017-0257-3.
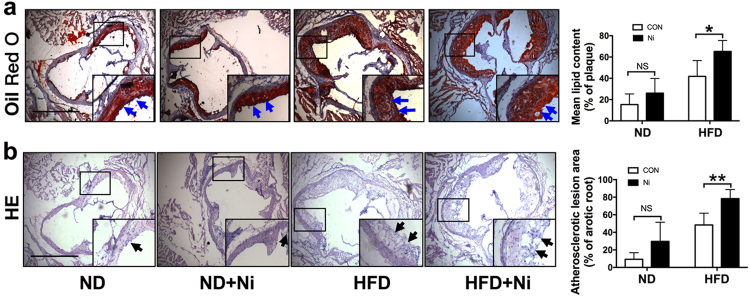
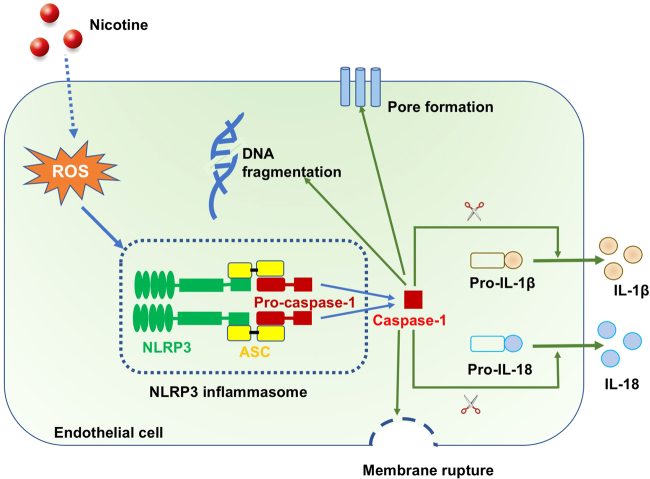
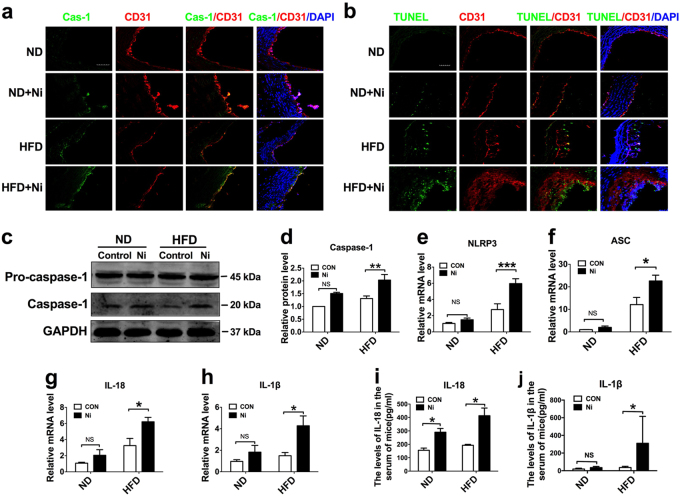
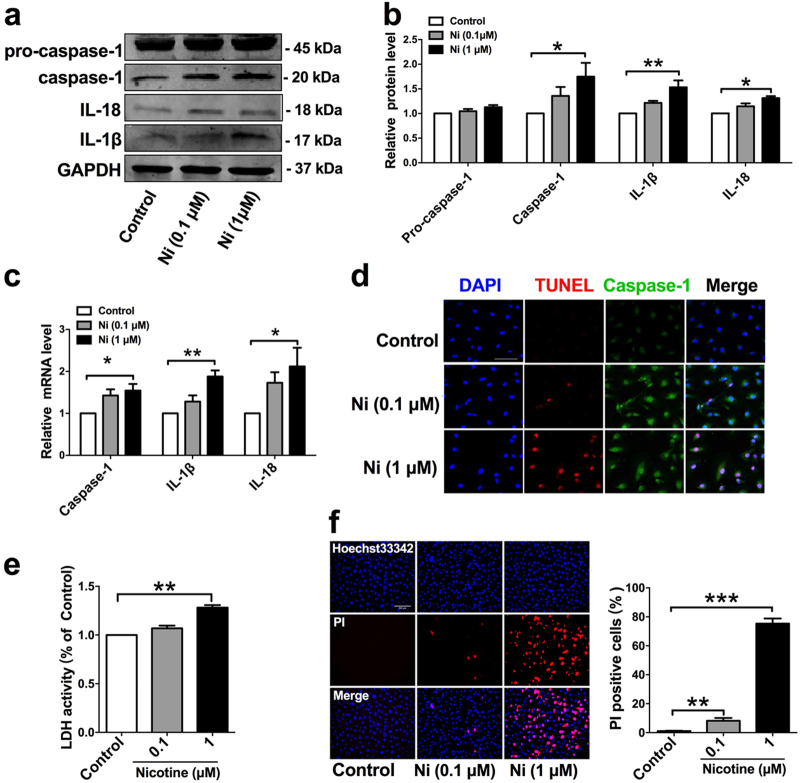
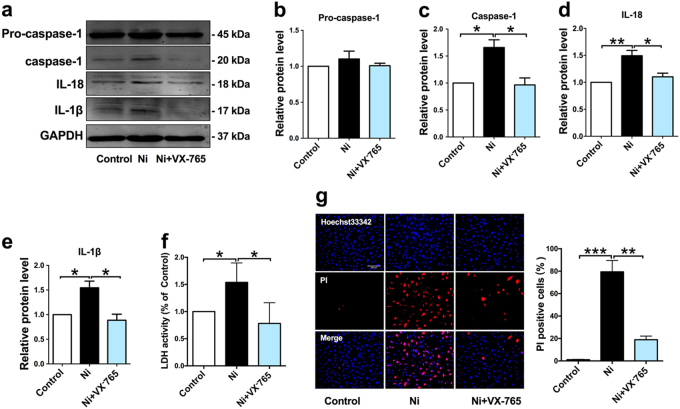
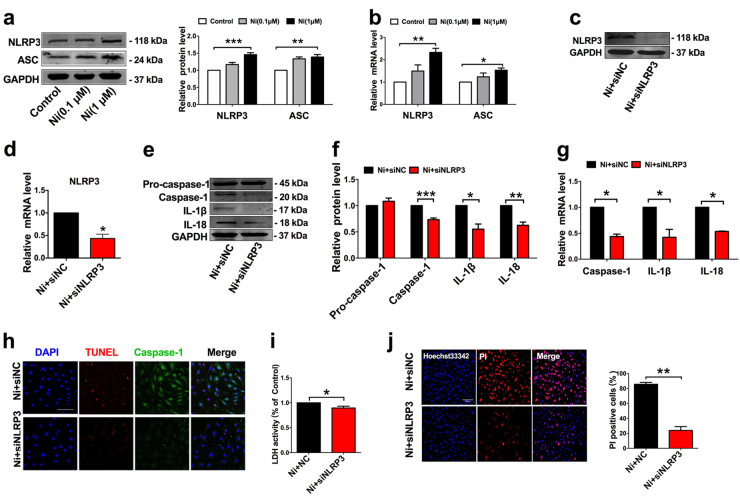
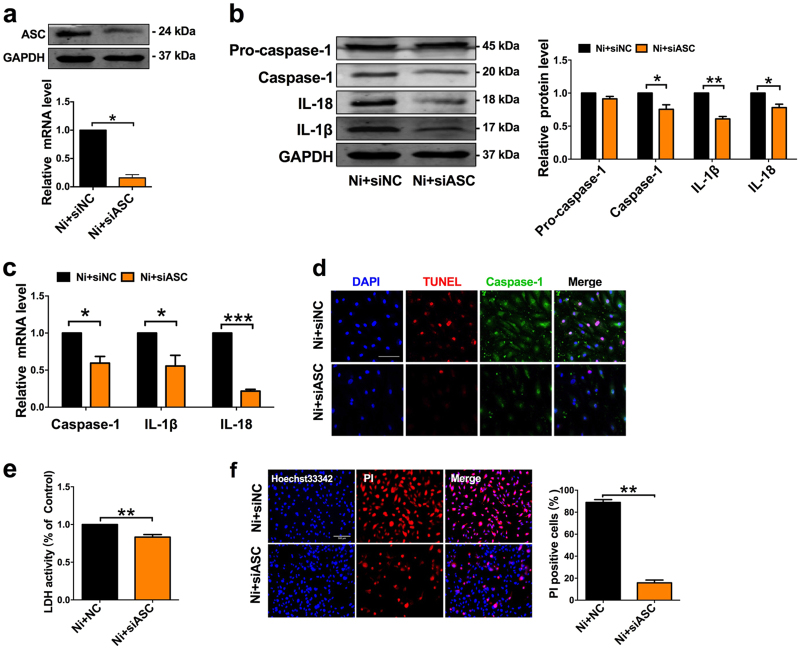
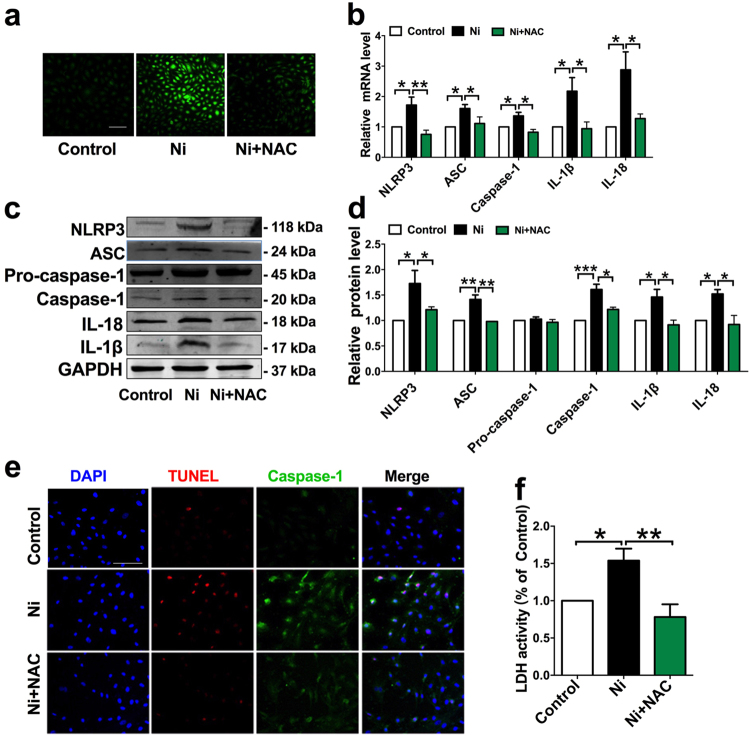
Cigarette smoking is a major risk factor for atherosclerosis and other cardiovascular diseases. Increasing evidence has demonstrated that nicotine impairs the cardiovascular system by targeting vascular endothelial cells, but the underlying mechanisms remain obscure. It is known that cell death and inflammation are crucial processes leading to atherosclerosis. We proposed that pyroptosis may be implicated in nicotine-induced atherosclerosis and therefore conducted the present study. We found that nicotine resulted in larger atherosclerotic plaques and secretion of inflammatory cytokines in ApoE mice fed with a high-fat diet (HFD). Treatment of human aortic endothelial cells (HAECs) with nicotine resulted in NLRP3-ASC inflammasome activation and pyroptosis, as evidenced by cleavage of caspase-1, production of downstream interleukin (IL)-1β and IL-18, and elevation of LDH activity and increase of propidium iodide (PI) positive cells, which were all inhibited by caspase-1 inhibitor. Moreover, silencing NLRP3 or ASC by small interfering RNA efficiently suppressed nicotine-induced caspase-1 cleavage, IL-18 and IL-1β production, and pyroptosis in HAECs. Further experiments revealed that the nicotine-NLRP3-ASC-pyroptosis pathway was activated by reactive oxygen species (ROS), since ROS scavenger (N-acetyl-cysteine, NAC) prevented endothelial cell pyroptosis. We conclude that pyroptosis is likely a cellular mechanism for the pro-atherosclerotic property of nicotine and stimulation of ROS to activate NLRP3 inflammasome is a signaling mechanism for nicotine-induced pyroptosis.
吸烟是动脉粥样硬化和其他心血管疾病的主要危险因素。越来越多的证据表明,尼古丁通过靶向血管内皮细胞损害心血管系统,但潜在机制尚不清楚。已知细胞死亡和炎症是导致动脉粥样硬化的关键过程。我们提出,细胞焦亡可能与尼古丁引起的动脉粥样硬化有关,因此进行了本研究。我们发现,尼古丁可导致载脂蛋白 E 基因敲除(ApoE)小鼠在高脂饮食喂养下形成更大的动脉粥样硬化斑块和分泌炎症细胞因子。用尼古丁处理人主动脉内皮细胞(HAEC)可导致 NLRP3-ASC 炎性小体激活和细胞焦亡,这表现在半胱氨酸天冬氨酸蛋白酶-1(caspase-1)的裂解、下游白细胞介素(IL)-1β和 IL-18 的产生以及乳酸脱氢酶(LDH)活性的升高和碘化丙啶(PI)阳性细胞的增加,这些均被 caspase-1 抑制剂所抑制。此外,小干扰 RNA 沉默 NLRP3 或 ASC 可有效抑制尼古丁诱导的 HAEC 中 caspase-1 的裂解、IL-18 和 IL-1β的产生以及细胞焦亡。进一步的实验表明,尼古丁-NLRP3-ASC 细胞焦亡途径被活性氧(ROS)激活,因为 ROS 清除剂(N-乙酰半胱氨酸,NAC)可防止内皮细胞发生细胞焦亡。我们的结论是,细胞焦亡可能是尼古丁促动脉粥样硬化作用的一种细胞机制,而 ROS 刺激激活 NLRP3 炎性小体是尼古丁诱导细胞焦亡的一种信号机制。