Yoshii Fumihito, Tomiyasu Hitoshi, Watanabe Ryo, Ryo Masafuchi
Department of Neurology, Saiseikai Shonan Hiratsuka Hospital, Hiratsuka, Japan.
Department of Neurology, Tokai University Oiso Hospital, Oiso, Japan.
Case Rep Neurol. 2017 Nov 10;9(3):267-271. doi: 10.1159/000481303. eCollection 2017 Sep-Dec.
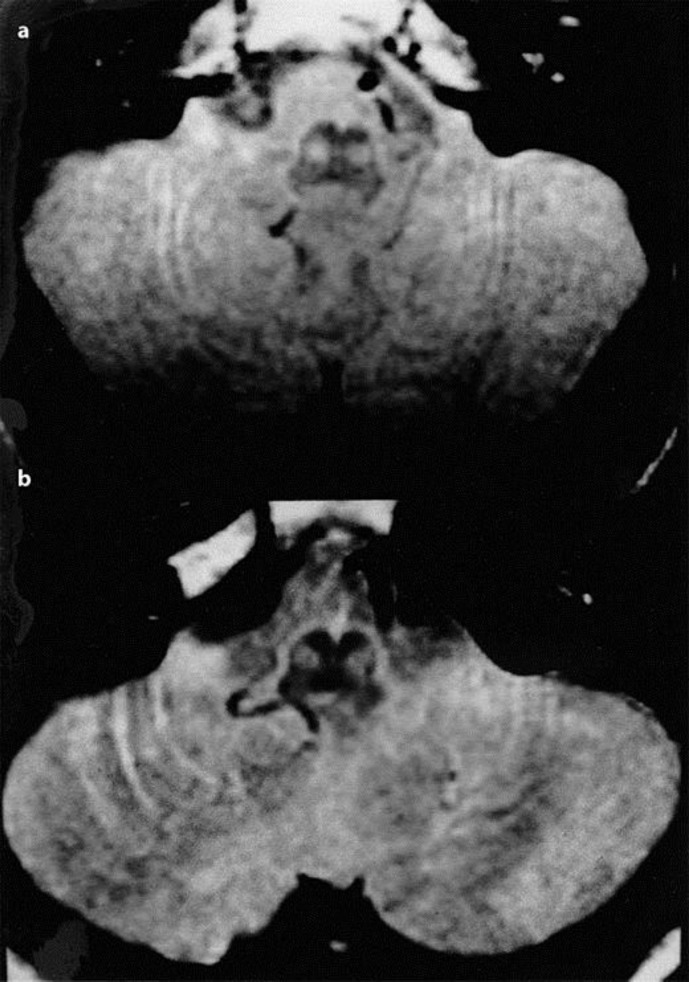
Spinocerebellar ataxia type 2 (SCA2) is an autosomal dominant spinocerebellar degeneration, associated with extended repeats of the trinucleotide CAG in the gene on the long arm of chromosome 12. Magnetic resonance imaging (MRI) of SCA2 showed significant atrophies of the brainstem, middle cerebellar peduncles, and cerebellum. We report two genetically proven SCA2 patients who showed hypertrophy of the inferior olivary nuclei on proton density- and T2-weighted MRI. This pattern has never been reported in patients with SCA1, SCA3, or SCA6, and may make it possible to differentiate SCA2 from other hereditary spinocerebellar ataxias.
2型脊髓小脑共济失调(SCA2)是一种常染色体显性脊髓小脑变性疾病,与12号染色体长臂上该基因中三核苷酸CAG的重复序列延长有关。SCA2的磁共振成像(MRI)显示脑干、小脑中脚和小脑有明显萎缩。我们报告了两名经基因证实的SCA2患者,他们在质子密度加权和T2加权MRI上显示下橄榄核肥大。这种模式在SCA1、SCA3或SCA6患者中从未有过报道,这可能有助于将SCA2与其他遗传性脊髓小脑共济失调区分开来。