Demir Eksi Durkadin, Shen Yiping, Erman Munire, Chorich Lynn P, Sullivan Megan E, Bilekdemir Meric, Yılmaz Elanur, Luleci Guven, Kim Hyung-Goo, Alper Ozgul M, Layman Lawrence C
Department of Medical Biology, Alanya Alaaddin Keykubat University, Faculty of Medicine, Antalya, Turkey.
2Guangxi Maternal and Child Health Hospital, Nanning, China.
Mol Cytogenet. 2018 Feb 3;11:13. doi: 10.1186/s13039-018-0359-3. eCollection 2018.
Little is known about the genetic contribution to Müllerian aplasia, better known to patients as Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Mutations in two genes ( and ) account for a small number of patients, but heterozygous copy number variants (CNVs) have been described. However, the significance of these CNVs in the pathogenesis of MRKH is unknown, but suggests possible autosomal dominant inheritance. We are not aware of CNV studies in consanguineous patients, which could pinpoint genes important in autosomal recessive MRKH. We therefore utilized SNP/CGH microarrays to identify CNVs and define regions of homozygosity (ROH) in Anatolian Turkish MRKH patients.
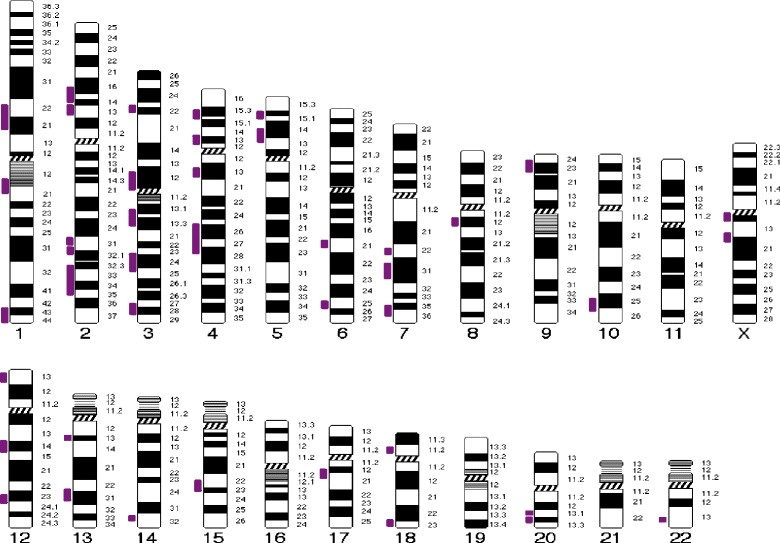
Five different CNVs were detected in 4/19 patients (21%), one of which is a previously reported 16p11.2 deletion containing 32 genes, while four involved smaller regions each containing only one gene. Fourteen of 19 (74%) of patients had parents that were third degree relatives or closer. There were 42 regions of homozygosity shared by at least two MRKH patients which was spread throughout most chromosomes. Of interest, eight candidate genes suggested by human or animal studies ( and ) reside within these ROH.
CNVs were found in about 20% of Turkish MRKH patients, and as in other studies, proof of causation is lacking. The 16p11.2 deletion seen in mixed populations is also identified in Turkish MRKH patients. Turkish MRKH patients have a higher likelihood of being consanguineous than the general Anatolian Turkish population. Although identified single gene mutations and heterozygous CNVs suggest autosomal dominant inheritance for MRKH in much of the western world, regions of homozygosity, which could contain shared mutant alleles, make it more likely that autosomal recessively inherited causes will be manifested in Turkish women with MRKH.
关于苗勒管发育不全(患者更熟知的是迈耶-罗基坦斯基-库斯特-豪泽综合征,即MRKH综合征)的遗传因素,人们了解甚少。两个基因的突变仅能解释少数患者的病因,但已有杂合拷贝数变异(CNV)的相关报道。然而,这些CNV在MRKH发病机制中的意义尚不清楚,但提示可能存在常染色体显性遗传。我们尚未发现针对近亲婚配患者的CNV研究,这类研究可能会找出在常染色体隐性遗传的MRKH中起重要作用的基因。因此,我们利用单核苷酸多态性/比较基因组杂交微阵列来识别CNV,并确定安纳托利亚土耳其MRKH患者的纯合区域(ROH)。
在19例患者中的4例(21%)检测到5种不同的CNV,其中一种是先前报道的16p11.2缺失,包含32个基因,而另外4种涉及较小区域,每个区域仅包含一个基因。19例患者中有14例(74%)的父母为三级亲属或更近的亲属关系。至少两名MRKH患者共有的纯合区域有42个,分布在大多数染色体上。有趣的是,人类或动物研究提示的8个候选基因(和)位于这些ROH内。
在约20%的土耳其MRKH患者中发现了CNV,与其他研究一样,缺乏因果关系的证据。在混合人群中发现的16p11.2缺失在土耳其MRKH患者中也有发现。土耳其MRKH患者近亲婚配的可能性高于一般安纳托利亚土耳其人群。尽管已识别出的单基因突变和杂合CNV提示在西方世界的大部分地区MRKH为常染色体显性遗传,但可能包含共享突变等位基因的纯合区域使得常染色体隐性遗传病因更有可能在患有MRKH的土耳其女性中表现出来。