Parente Valeria, Corti Stefania
Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Via Francesco Sforza 35, 20122 Milan, Italy.
Ther Adv Neurol Disord. 2018 Feb 5;11:1756285618754501. doi: 10.1177/1756285618754501. eCollection 2018.
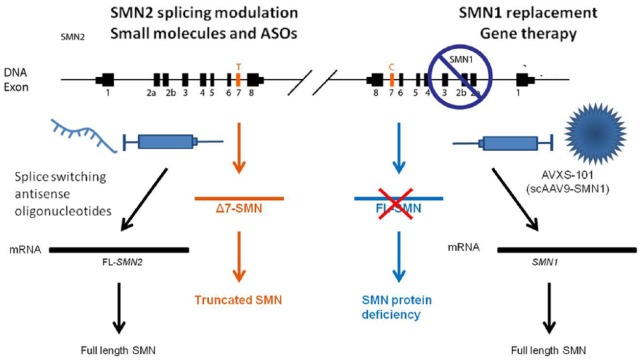
Spinal muscular atrophy (SMA) is a progressive, recessively inherited neuromuscular disease, characterized by the degeneration of lower motor neurons in the spinal cord and brainstem, which leads to weakness and muscle atrophy. SMA currently represents the most common genetic cause of infant death. SMA is caused by the lack of survival motor neuron (SMN) protein due to mutations, which are often deletions, in the gene. In the absence of treatments able to modify the disease course, a considerable burden falls on patients and their families. Greater knowledge of the molecular basis of SMA pathogenesis has fuelled the development of potential therapeutic approaches, which are illustrated here. Nusinersen, a modified antisense oligonucleotide that modulates the splicing of the mRNA transcript, is the first approved drug for all types of SMA. Moreover, the first gene therapy clinical trial using adeno-associated virus (AAV) vectors encoding SMN reported positive results in survival and motor milestones achievement. In addition, other strategies are in the pipeline, including modulation of transcripts, neuroprotection, and targeting an increasing number of other peripheral targets, including the skeletal muscle. Based on this premise, it is reasonable to expect that therapeutic approaches aimed at treating SMA will soon be changed, and improved, in a meaningful way. We discuss the challenges with regard to the development of novel treatments for patients with SMA, and depict the current and future scenarios as the field enters into a new era of promising effective treatments.
脊髓性肌萎缩症(SMA)是一种进行性隐性遗传神经肌肉疾病,其特征为脊髓和脑干中的下运动神经元退化,进而导致肌无力和肌肉萎缩。SMA目前是婴儿死亡最常见的遗传原因。SMA是由于基因发生突变(通常为缺失)导致存活运动神经元(SMN)蛋白缺乏所致。在缺乏能够改变疾病进程的治疗方法的情况下,患者及其家庭承受着相当大的负担。对SMA发病机制分子基础的更多了解推动了潜在治疗方法的发展,本文对此进行了阐述。诺西那生钠是一种修饰的反义寡核苷酸,可调节mRNA转录本的剪接,是首个获批用于所有类型SMA的药物。此外,首个使用编码SMN的腺相关病毒(AAV)载体进行的基因治疗临床试验在生存和运动里程碑达成方面报告了阳性结果。此外,其他策略也在筹备中,包括调节转录本、神经保护以及针对越来越多的其他外周靶点(包括骨骼肌)。基于这一前提,有理由期待旨在治疗SMA的治疗方法很快会以有意义的方式得到改变和改进。我们讨论了为SMA患者开发新疗法所面临的挑战,并描绘了随着该领域进入有前景的有效治疗新时代的当前和未来情景。