Cai Xiaotang, Yu Dan, Xie Yongmei, Zhou Hui
Department of Pediatrics, West China Second University Hospital Key Laboratory of Obstetric and Gynaecologic and Pediatric Diseases and Birth Defects of Ministry of Education, Sichuan University, Chengdu, Sichuan, China.
Medicine (Baltimore). 2018 Feb;97(7):e9880. doi: 10.1097/MD.0000000000009880.
Argininemia is an autosomal recessive inherited disorder of the urea cycle. Because of its atypical symptoms in early age, diagnosis can be delayed until the typical chronic manifestations - including spastic diplegia, deterioration in cognitive function, and epilepsy - appear in later childhood.
A Chinese boy initially presented with severe stunting and partial growth hormone deficiency (PGHD) at 3 years old and was initially treated with growth hormone replacement therapy. Seven years later (at 10 years old), he presented with spastic diplegia, cognitive function lesions, epilepsy, and peripheral neuropathy.
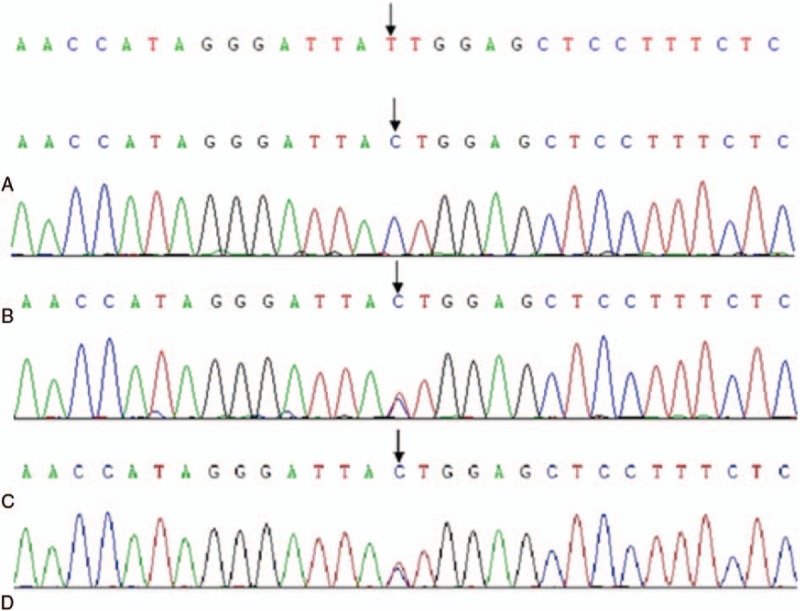
Ultimately, the patient was diagnosed with argininemia with homozygous mutation (c.32T>C) of the ARG1 gene at 10 years old. Blood tests showed mildly elevated blood ammonia and creatine kinase, and persistently elevated bilirubin.
Protein intake was limited to 0.8 g/kg/day, citrulline (150-200 mg [kg d]) was prescribed.
The patient's mental state and vomiting had improved after 3 months treatment. At 10 years and 9 month old, his height and weight had reached 121cm and 22kg, respectively, but his spastic diplegia symptoms had not improved.
This case demonstrates that stunting and PGHD that does not respond to growth hormone replacement therapy might hint at inborn errors of metabolism (IEM). IEM should also be considered in patients with persistently elevated bilirubin with or without abnormal liver transaminase, as well as elevated blood ammonia and creatine kinase, in the absence of hepatic disease.
精氨酸血症是一种常染色体隐性遗传的尿素循环障碍疾病。因其在幼年时症状不典型,诊断可能会延迟,直到典型的慢性表现(包括痉挛性双侧瘫、认知功能减退和癫痫)在儿童后期出现。
一名中国男孩3岁时最初表现为严重发育迟缓及部分生长激素缺乏(PGHD),最初接受生长激素替代治疗。7年后(10岁时),他出现痉挛性双侧瘫、认知功能损害、癫痫和周围神经病变。
最终,该患者在10岁时被诊断为精氨酸血症,伴有ARG1基因纯合突变(c.32T>C)。血液检查显示血氨和肌酸激酶轻度升高,胆红素持续升高。
蛋白质摄入量限制为0.8克/(千克·天),并开具了瓜氨酸(150 - 200毫克/[千克·天])。
治疗3个月后患者的精神状态和呕吐情况有所改善。在10岁9个月时,他的身高和体重分别达到了121厘米和22千克,但痉挛性双侧瘫症状没有改善。
该病例表明,对生长激素替代治疗无反应的发育迟缓和PGHD可能提示先天性代谢缺陷(IEM)。对于胆红素持续升高(无论有无肝转氨酶异常)以及血氨和肌酸激酶升高且无肝脏疾病的患者,也应考虑IEM。