Gray David L, Allen John A, Mente Scot, O'Connor Rebecca E, DeMarco George J, Efremov Ivan, Tierney Patrick, Volfson Dmitri, Davoren Jennifer, Guilmette Edward, Salafia Michelle, Kozak Rouba, Ehlers Michael D
Medicine Design, Pfizer Worldwide Research & Development, Cambridge, MA, 02139, USA.
Internal Medicine, Pfizer Worldwide Research & Development, Cambridge, MA, 02139, USA.
Nat Commun. 2018 Feb 14;9(1):674. doi: 10.1038/s41467-017-02776-7.
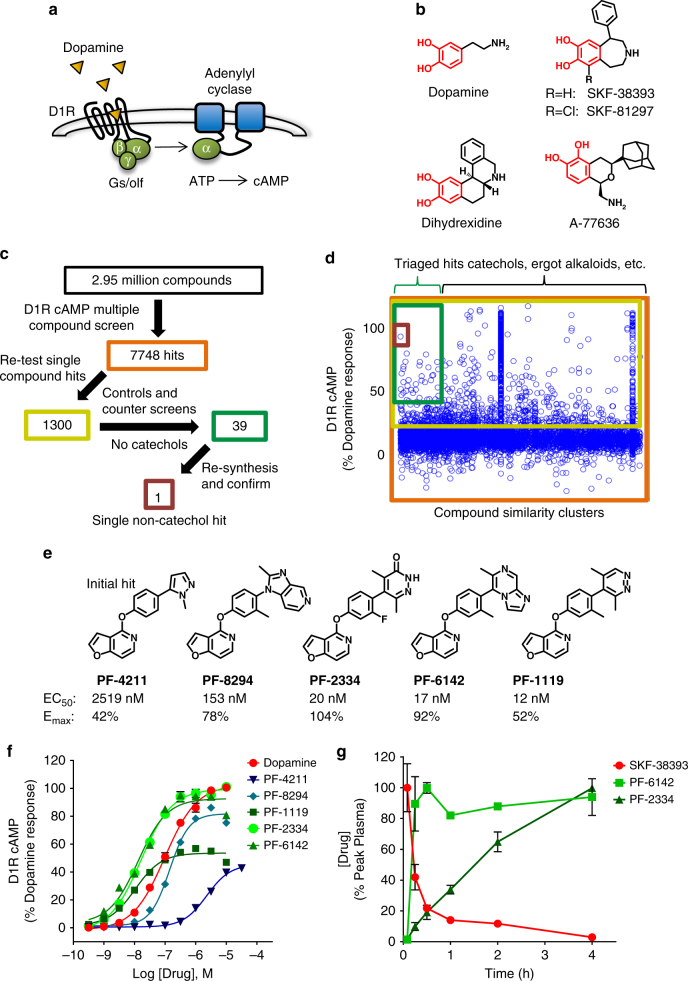
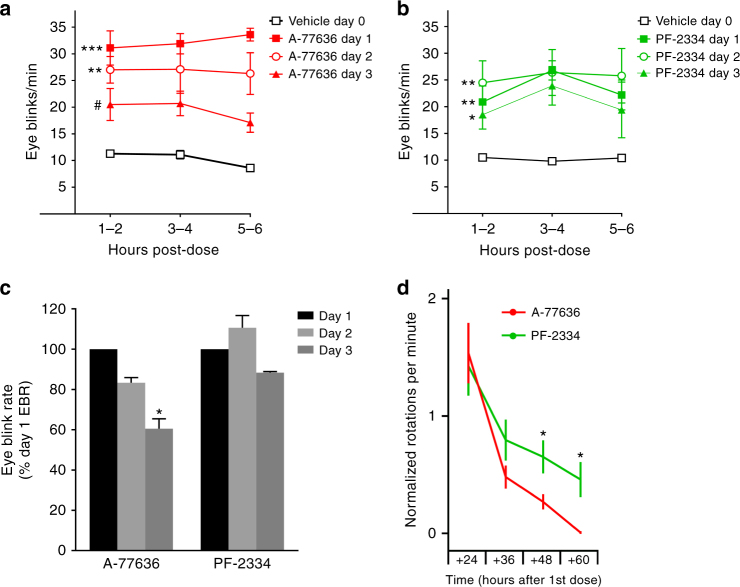
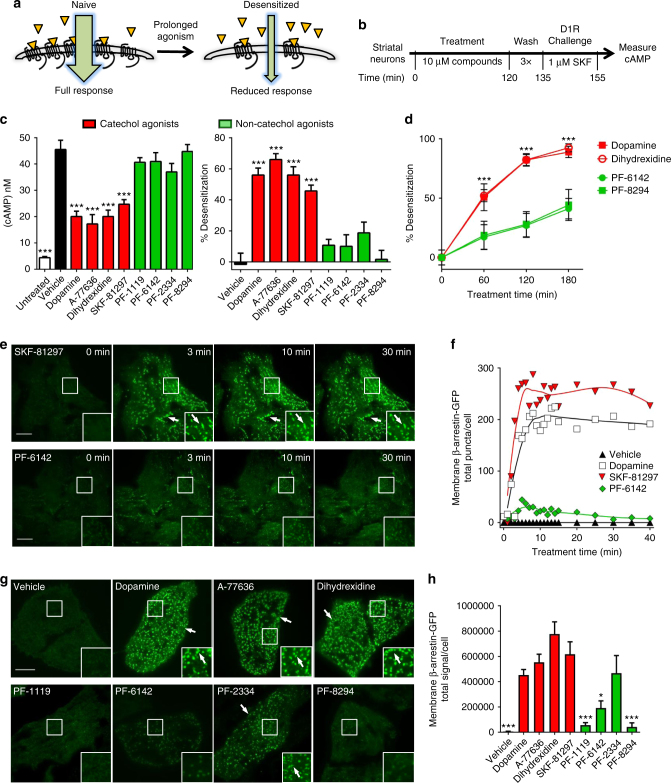
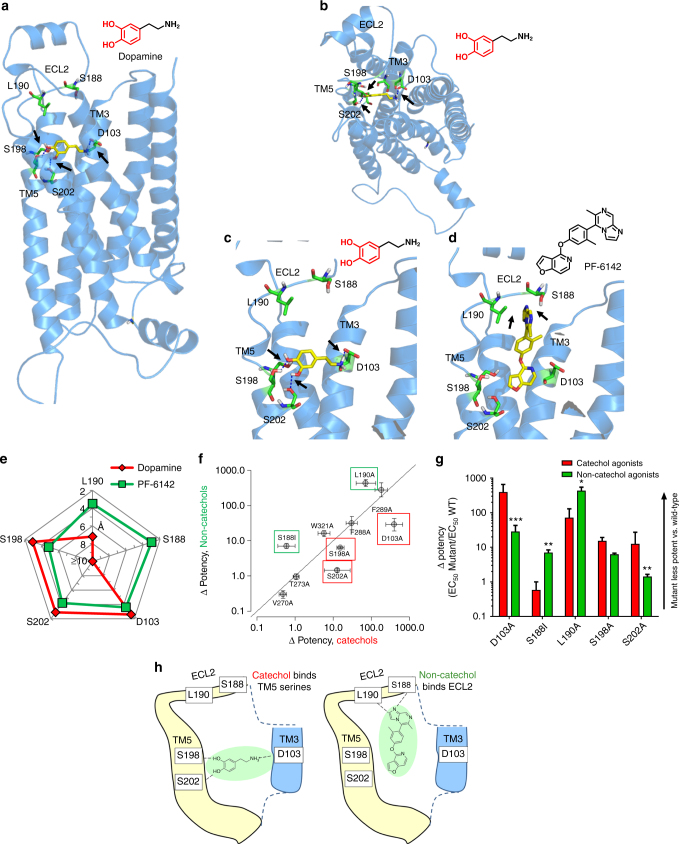
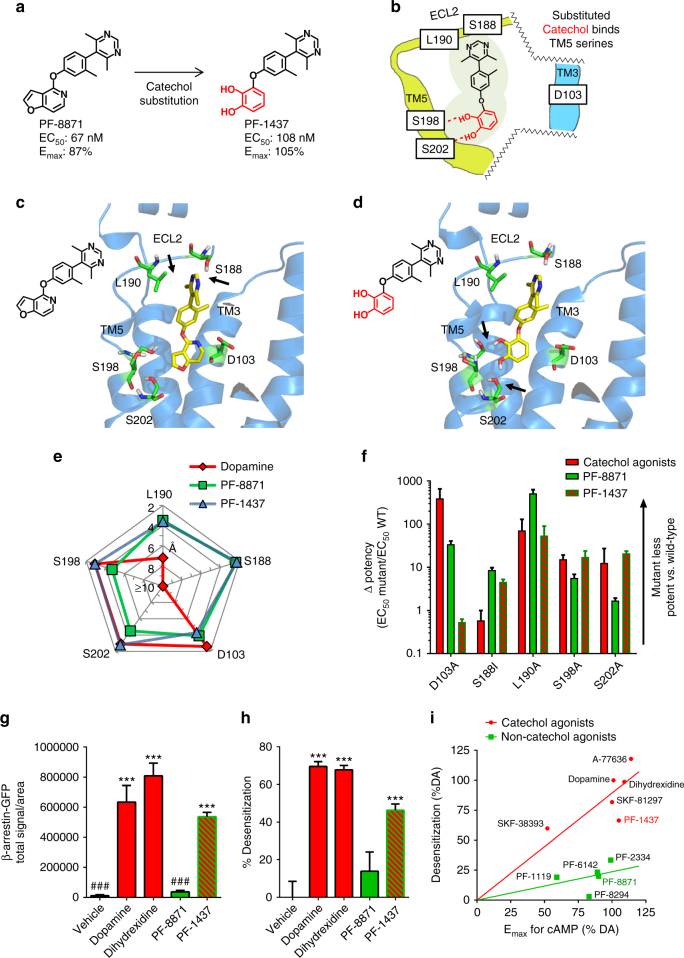
Selective activation of dopamine D1 receptors (D1Rs) has been pursued for 40 years as a therapeutic strategy for neurologic and psychiatric diseases due to the fundamental role of D1Rs in motor function, reward processing, and cognition. All known D1R-selective agonists are catechols, which are rapidly metabolized and desensitize the D1R after prolonged exposure, reducing agonist response. As such, drug-like selective D1R agonists have remained elusive. Here we report a novel series of selective, potent non-catechol D1R agonists with promising in vivo pharmacokinetic properties. These ligands stimulate adenylyl cyclase signaling and are efficacious in a rodent model of Parkinson's disease after oral administration. They exhibit distinct binding to the D1R orthosteric site and a novel functional profile including minimal receptor desensitization, reduced recruitment of β-arrestin, and sustained in vivo efficacy. These results reveal a novel class of D1 agonists with favorable drug-like properties, and define the molecular basis for catechol-specific recruitment of β-arrestin to D1Rs.
由于多巴胺D1受体(D1Rs)在运动功能、奖赏处理和认知中发挥着重要作用,40年来,人们一直致力于将选择性激活D1Rs作为治疗神经和精神疾病的一种策略。所有已知的D1R选择性激动剂都是儿茶酚类化合物,它们在长时间暴露后会迅速代谢并使D1R脱敏,从而降低激动剂反应。因此,类似药物的选择性D1R激动剂一直难以获得。在此,我们报告了一系列新型的选择性、强效非儿茶酚D1R激动剂,它们具有良好的体内药代动力学特性。这些配体刺激腺苷酸环化酶信号传导,口服给药后在帕金森病啮齿动物模型中有效。它们与D1R正构位点表现出独特的结合以及一种新型功能特征,包括最小化的受体脱敏、β-抑制蛋白募集减少和持续的体内疗效。这些结果揭示了一类具有良好药物样特性的新型D1激动剂,并确定了儿茶酚特异性募集β-抑制蛋白至D1Rs的分子基础。