Mathieson Toby, Franken Holger, Kosinski Jan, Kurzawa Nils, Zinn Nico, Sweetman Gavain, Poeckel Daniel, Ratnu Vikram S, Schramm Maike, Becher Isabelle, Steidel Michael, Noh Kyung-Min, Bergamini Giovanna, Beck Martin, Bantscheff Marcus, Savitski Mikhail M
Cellzome GmbH, GlaxoSmithKline, Meyerhofstraße 1, 69117, Heidelberg, Germany.
Structural and Computational Biology Unit, European Molecular Biology Laboratory, Meyerhofstraße 1, 69117, Heidelberg, Germany.
Nat Commun. 2018 Feb 15;9(1):689. doi: 10.1038/s41467-018-03106-1.
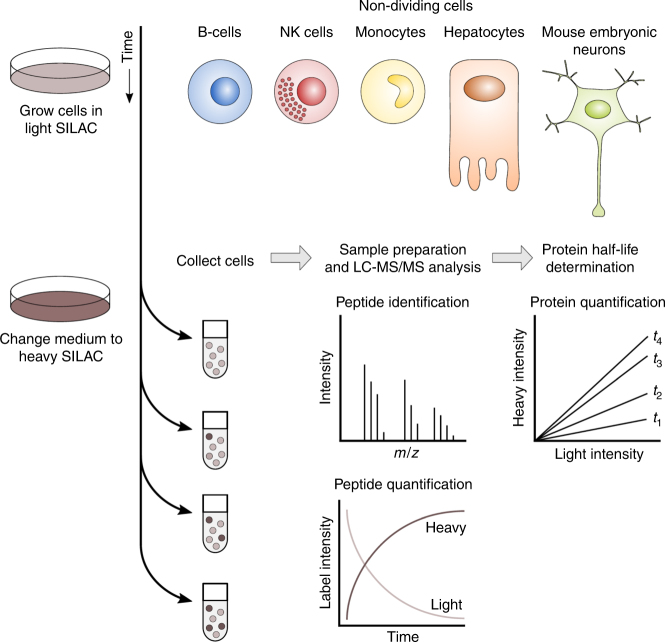
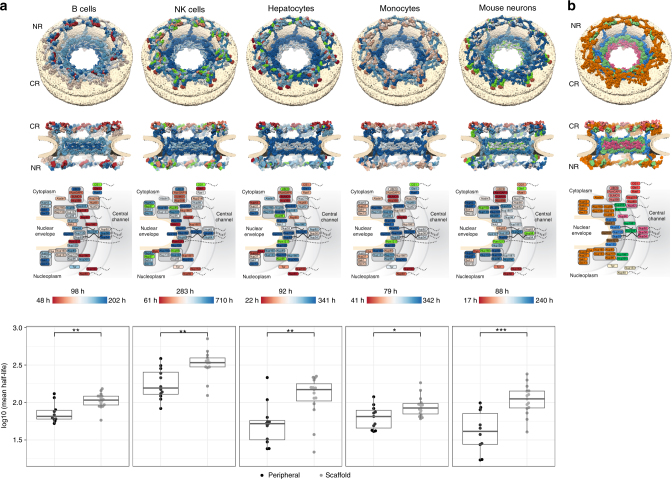
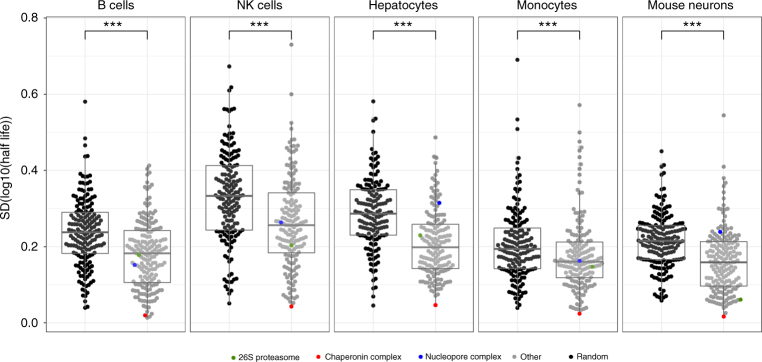
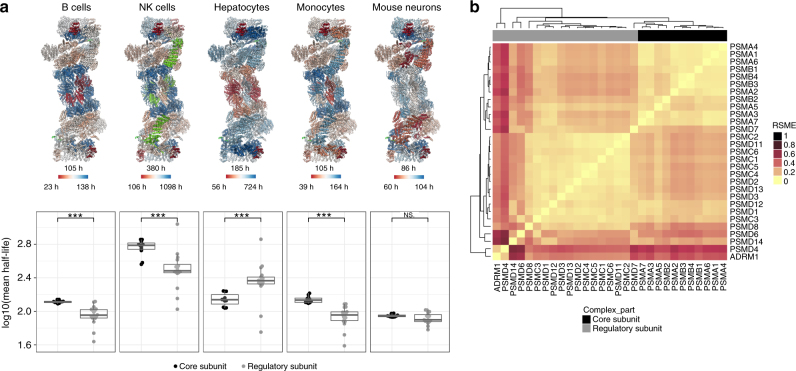
A better understanding of proteostasis in health and disease requires robust methods to determine protein half-lives. Here we improve the precision and accuracy of peptide ion intensity-based quantification, enabling more accurate protein turnover determination in non-dividing cells by dynamic SILAC-based proteomics. This approach allows exact determination of protein half-lives ranging from 10 to >1000 h. We identified 4000-6000 proteins in several non-dividing cell types, corresponding to 9699 unique protein identifications over the entire data set. We observed similar protein half-lives in B-cells, natural killer cells and monocytes, whereas hepatocytes and mouse embryonic neurons show substantial differences. Our data set extends and statistically validates the previous observation that subunits of protein complexes tend to have coherent turnover. Moreover, analysis of different proteasome and nuclear pore complex assemblies suggests that their turnover rate is architecture dependent. These results illustrate that our approach allows investigating protein turnover and its implications in various cell types.
要更好地理解健康与疾病状态下的蛋白质稳态,需要有可靠的方法来测定蛋白质半衰期。在此,我们提高了基于肽离子强度定量的精度和准确性,通过基于动态稳定同位素标记氨基酸细胞培养法(SILAC)的蛋白质组学,能够更准确地测定非分裂细胞中的蛋白质周转率。这种方法可以精确测定10至1000多小时的蛋白质半衰期。我们在几种非分裂细胞类型中鉴定出4000 - 6000种蛋白质,在整个数据集中对应9699个独特的蛋白质鉴定结果。我们观察到B细胞、自然杀伤细胞和单核细胞中的蛋白质半衰期相似,而肝细胞和小鼠胚胎神经元则有显著差异。我们的数据集扩展并从统计学上验证了先前的观察结果,即蛋白质复合物的亚基往往具有一致的周转率。此外,对不同蛋白酶体和核孔复合体组件的分析表明,它们的周转率取决于结构。这些结果表明,我们的方法能够研究各种细胞类型中的蛋白质周转及其影响。