Sex Transm Dis. 2018 Apr;45(4):222-228. doi: 10.1097/OLQ.0000000000000726.
The integration of traditional contact tracing with HIV sequence analyses offers opportunities to mitigate some of the barriers to effective network construction. We used combined analyses during an outbreak investigation of spatiotemporally clustered acute HIV infections to evaluate if the observed clustering was the product of a single outbreak.
We investigated acute and recent HIV index cases reported in North Carolina from 2013 to 2014 and their reported contacts. Contact tracing networks were constructed with surveillance data and compared with phylogenetic transmission clusters involving an index case using available HIV-1 pol sequences including 1672 references. Clusters were defined as clades of 2 or more sequences with a less than 1.5% genetic distance and a bootstrap of at least 98% on maximum-likelihood phylogenies.
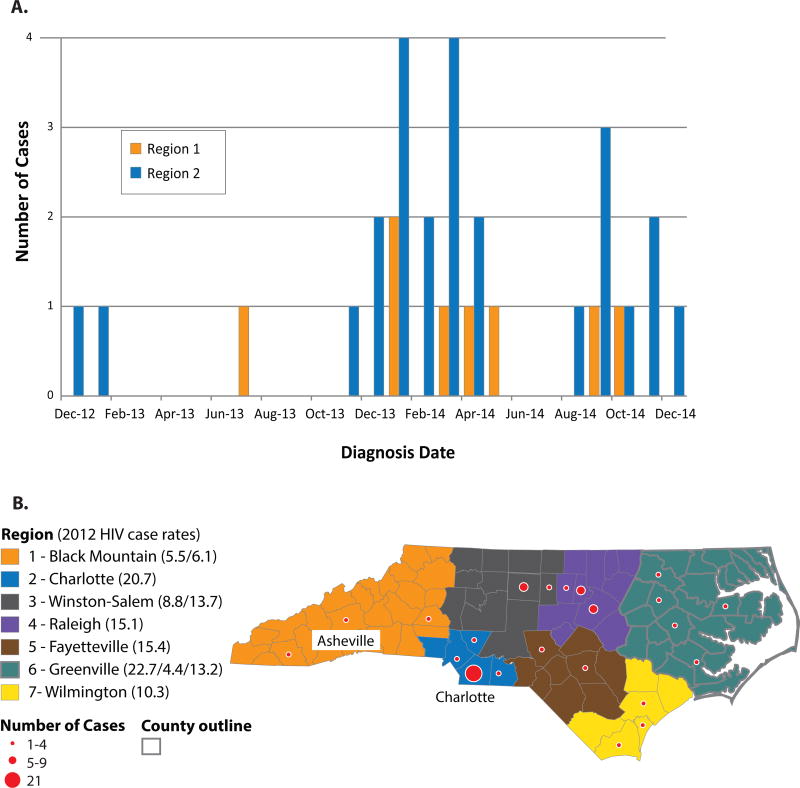
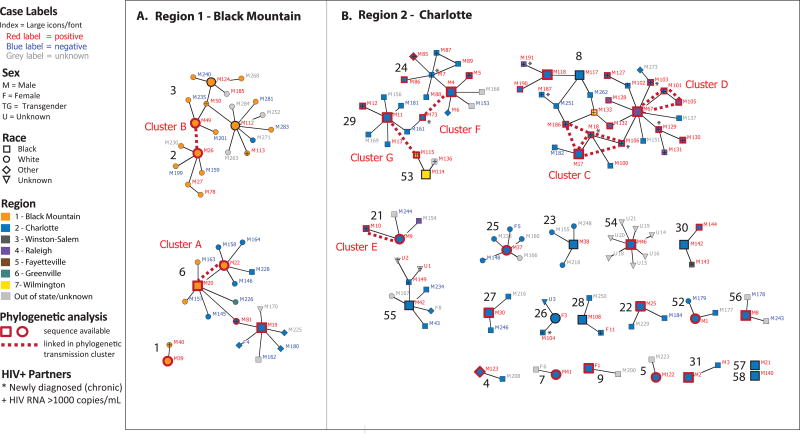
In total, 68 index cases and 210 contacts (71 HIV infected) were reported. The contact tracing network involved 58 components with low overall density (1.2% statewide); 33% of first-degree contacts could not be located. Among 38 (56%) of 68 index cases and 34 (48%) of 71 contacts with sequences, 13 phylogenetic clusters were identified (size 2-4 members). Four clusters connected network components that were not linked in contact tracing. The largest component (n = 28 cases) included 2 distinct phylogenetic clusters and spanned 2 regions.
We identified the concurrent expansion of multiple small transmission clusters rather than a single outbreak in a largely disconnected contact tracing network. Integration of phylogenetic analyses provided timely information on transmission networks during the investigation. Our findings highlight the potential of combined methods to better identify high-risk networks for intervention.
将传统的接触者追踪与 HIV 序列分析相结合,为克服有效网络构建的一些障碍提供了机会。我们在对时空聚集性急性 HIV 感染爆发的调查中,使用联合分析来评估所观察到的聚集是否是单一爆发的产物。
我们调查了 2013 年至 2014 年在北卡罗来纳州报告的急性和近期 HIV 索引病例及其报告的接触者。使用监测数据构建了接触者追踪网络,并将其与涉及索引病例的系统发育传播簇进行比较,使用了包括 1672 个参考序列在内的可用 HIV-1 pol 序列。簇定义为具有小于 1.5%遗传距离和最大似然系统发育上至少 98%的自举值的 2 个或更多序列的分支。
总共报告了 68 例索引病例和 210 名接触者(71 例 HIV 感染)。接触者追踪网络涉及 58 个组件,整体密度较低(全州 1.2%);33%的一级接触者无法找到。在 68 例索引病例中的 38 例(56%)和 71 例有序列的接触者中的 34 例(48%)中,确定了 13 个系统发育簇(大小为 2-4 个成员)。4 个簇连接了接触者追踪中未连接的网络组件。最大的组件(n=28 例)包括 2 个不同的系统发育簇,跨越 2 个地区。
我们发现,在一个基本不相连的接触者追踪网络中,多个小传播簇同时扩张,而不是单一爆发。系统发育分析的整合在调查期间及时提供了关于传播网络的信息。我们的研究结果强调了联合方法在识别高危干预网络方面的潜力。