Gaucher Clinic, Shaare Zedek Medical Center, Hebrew University and Hadassah Medical School, 12 Bayit Street, P.O. Box 3235, 91031, Jerusalem, Israel.
Pfizer Inc, New York, NY, USA.
Orphanet J Rare Dis. 2018 Feb 23;13(1):36. doi: 10.1186/s13023-018-0776-8.
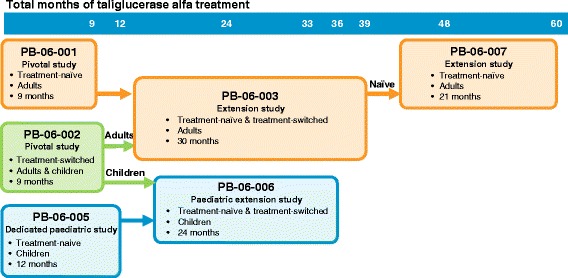
Taliglucerase alfa is an enzyme replacement therapy (ERT) approved for treatment of adult and paediatric patients with Type 1 Gaucher disease (GD) in several countries and the first plant cell-expressed recombinant therapeutic protein approved by the US Food and Drug Administration for humans. Here, we review the findings across six key taliglucerase alfa clinical studies. A total of 33 treatment-naïve adult patients were randomized to taliglucerase alfa 30 U/kg or 60 U/kg in a 9-month, multicentre, randomized, double-blind, parallel-group, dose-comparison pivotal study, after which eligible patients continued into two consecutive extension studies; 17 treatment-naïve adult patients completed 5 total years of treatment with taliglucerase alfa. In the only ERT study focused on exclusively paediatric patients with GD, 11 treatment-naïve children were randomized to taliglucerase alfa 30 U/kg or 60 U/kg in a 12-month, multicentre, double-blind study; nine completed 3 total years of treatment in a dedicated paediatric extension study. The effect of switching patients from imiglucerase to taliglucerase alfa was also investigated in a separate 9-month study that included 26 adults and five children; 10 adults completed a total of 3 years and two children completed a total of 2.75 years of taliglucerase alfa treatment in the extension studies. All studies evaluated safety and spleen volume, liver volume, platelet count, haemoglobin concentration, and biomarkers as measures of efficacy. Detailed results from baseline through the end of these studies are presented. Taliglucerase alfa was well tolerated, and adverse events were generally mild/moderate in severity and transient. Treatment with taliglucerase alfa resulted in improvements (treatment-naïve patients) or stability (patients switched from imiglucerase) in visceral, haematologic, and biomarker parameters. Together, this comprehensive data set supports the treatment of adult and paediatric patients with GD who are naïve to ERT or who have previously been treated with imiglucerase.
替利葡萄糖脑苷脂酶 α 是一种酶替代疗法 (ERT),已在多个国家获得批准,用于治疗 1 型 Gaucher 病 (GD) 的成人和儿科患者,也是美国食品和药物管理局批准的首个用于人类的植物细胞表达的重组治疗蛋白。在这里,我们回顾了六项关键的替利葡萄糖脑苷脂酶 α 临床研究结果。共有 33 名未经治疗的成年患者在一项为期 9 个月、多中心、随机、双盲、平行组、剂量比较的关键研究中随机分配至替利葡萄糖脑苷脂酶 α 30 U/kg 或 60 U/kg,之后符合条件的患者继续进入两项连续的扩展研究;17 名未经治疗的成年患者总共接受了 5 年的替利葡萄糖脑苷脂酶 α 治疗。在唯一一项专门针对 GD 儿科患者的 ERT 研究中,11 名未经治疗的儿童在一项为期 12 个月的多中心、双盲研究中随机分配至替利葡萄糖脑苷脂酶 α 30 U/kg 或 60 U/kg;9 名儿童在专门的儿科扩展研究中完成了 3 年的治疗。在另一项为期 9 个月的研究中,还研究了将患者从伊米葡萄糖脑苷脂酶转换为替利葡萄糖脑苷脂酶 α 的效果,该研究包括 26 名成年患者和 5 名儿童;10 名成年患者总共完成了 3 年的治疗,2 名儿童在扩展研究中总共完成了 2.75 年的替利葡萄糖脑苷脂酶 α 治疗。所有研究均评估了安全性和脾体积、肝体积、血小板计数、血红蛋白浓度和生物标志物作为疗效的衡量标准。本文呈现了从基线到这些研究结束的详细结果。替利葡萄糖脑苷脂酶 α 具有良好的耐受性,不良事件通常为轻度/中度且为一过性。替利葡萄糖脑苷脂酶 α 的治疗可改善(未经治疗的患者)或稳定(曾接受伊米葡萄糖脑苷脂酶治疗的患者)内脏、血液学和生物标志物参数。总的来说,这组综合数据集支持对 ERT 初治或曾接受伊米葡萄糖脑苷脂酶治疗的 GD 成人和儿科患者进行治疗。