Lamarre Sophie, Frasse Pierre, Zouine Mohamed, Labourdette Delphine, Sainderichin Elise, Hu Guojian, Le Berre-Anton Véronique, Bouzayen Mondher, Maza Elie
LISBP, Centre National de la Recherche Scientifique, INRA, INSA, Université de Toulouse, Toulouse, France.
GBF, Université de Toulouse, INRA, Castanet-Tolosan, France.
Front Plant Sci. 2018 Feb 14;9:108. doi: 10.3389/fpls.2018.00108. eCollection 2018.
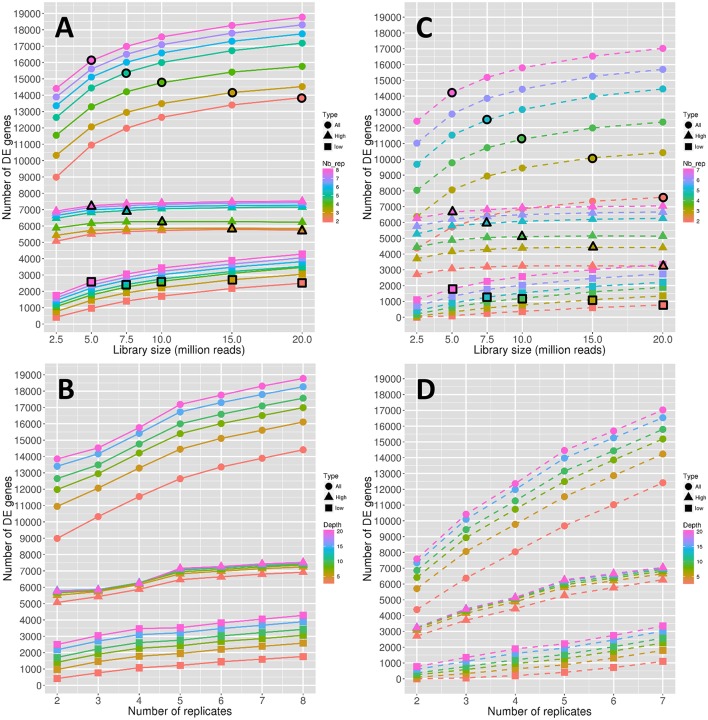
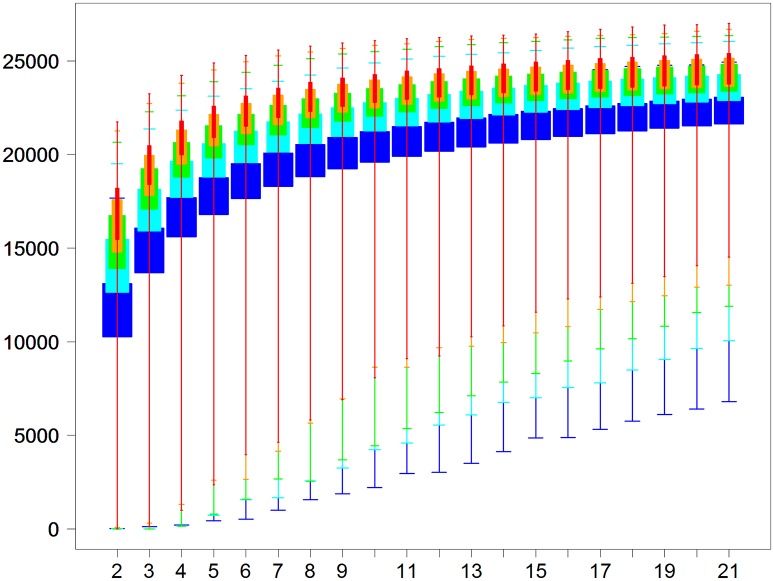
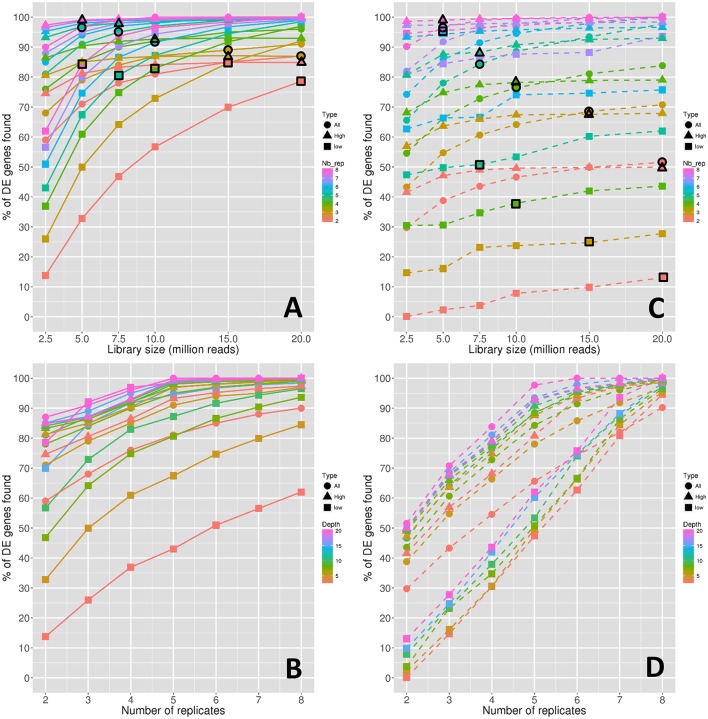
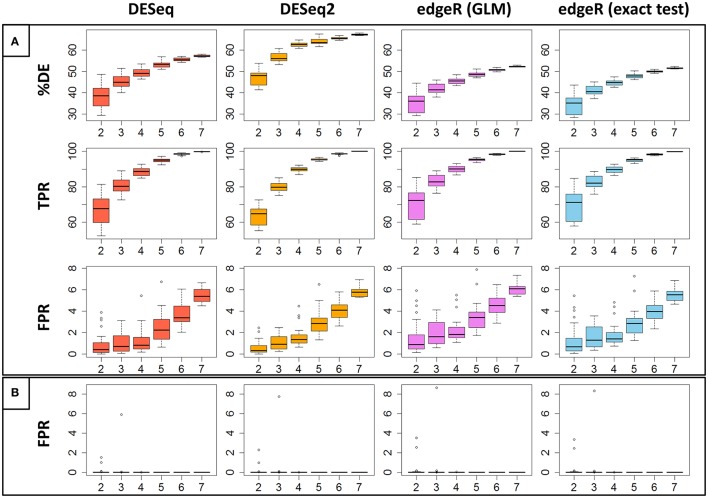
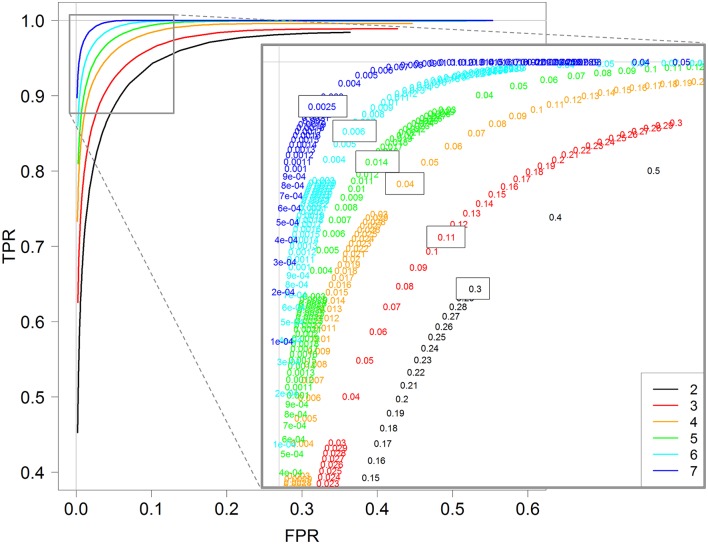
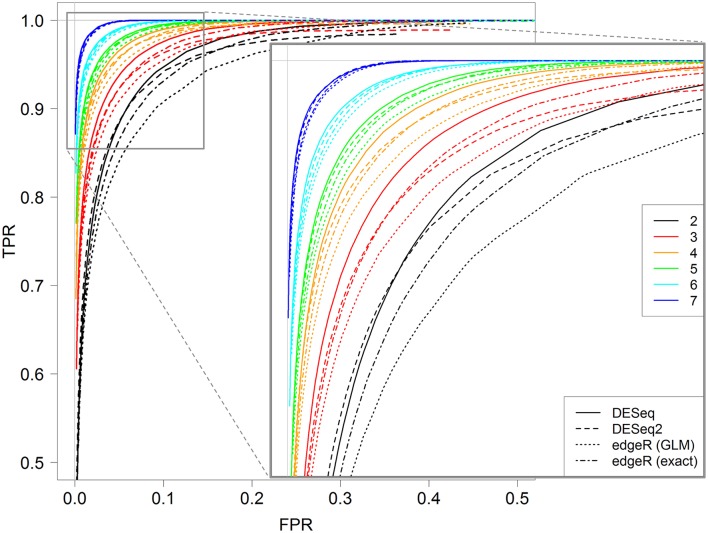
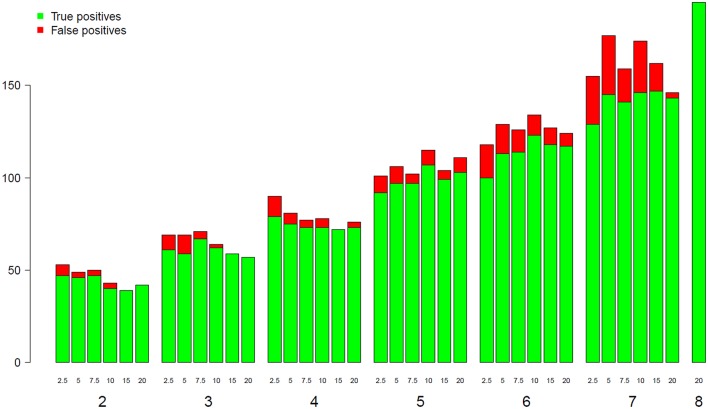
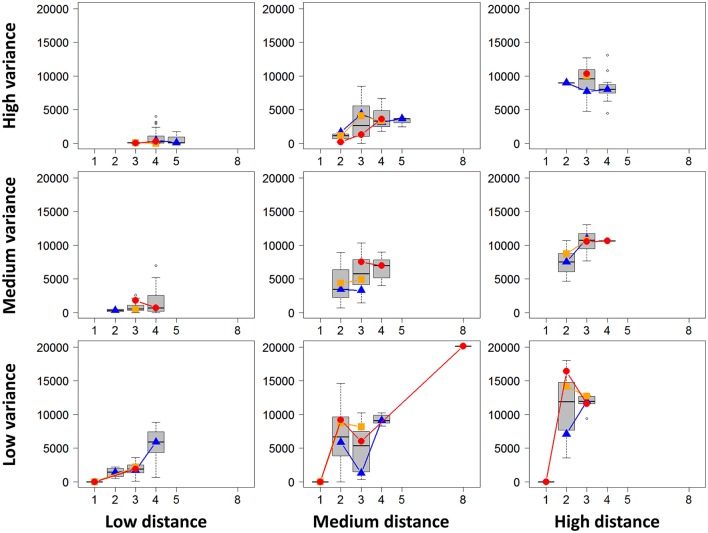
RNA-Seq is a widely used technology that allows an efficient genome-wide quantification of gene expressions for, for example, differential expression (DE) analysis. After a brief review of the main issues, methods and tools related to the DE analysis of RNA-Seq data, this article focuses on the impact of both the replicate number and library size in such analyses. While the main drawback of previous relevant studies is the lack of generality, we conducted both an analysis of a two-condition experiment (with eight biological replicates per condition) to compare the results with previous benchmark studies, and a meta-analysis of 17 experiments with up to 18 biological conditions, eight biological replicates and 100 million (M) reads per sample. As a global trend, we concluded that the replicate number has a larger impact than the library size on the power of the DE analysis, except for low-expressed genes, for which both parameters seem to have the same impact. Our study also provides new insights for practitioners aiming to enhance their experimental designs. For instance, by analyzing both the sensitivity and specificity of the DE analysis, we showed that the optimal threshold to control the false discovery rate (FDR) is approximately 2, where r is the replicate number. Furthermore, we showed that the false positive rate (FPR) is rather well controlled by all three studied R packages: , and . We also analyzed the impact of both the replicate number and library size on gene ontology (GO) enrichment analysis. Interestingly, we concluded that increases in the replicate number and library size tend to enhance the sensitivity and specificity, respectively, of the GO analysis. Finally, we recommend to RNA-Seq practitioners the production of a pilot data set to strictly analyze the power of their experimental design, or the use of a public data set, which should be similar to the data set they will obtain. For individuals working on tomato research, on the basis of the meta-analysis, we recommend at least four biological replicates per condition and 20 M reads per sample to be almost sure of obtaining about 1000 DE genes if they exist.
RNA测序是一种广泛应用的技术,它能够对全基因组范围内的基因表达进行高效定量分析,例如用于差异表达(DE)分析。在简要回顾了与RNA测序数据DE分析相关的主要问题、方法和工具之后,本文重点关注此类分析中重复样本数量和文库大小的影响。虽然先前相关研究的主要缺点是缺乏普遍性,但我们既进行了双条件实验分析(每个条件有8个生物学重复),以便将结果与先前的基准研究进行比较,又对17个实验进行了荟萃分析,这些实验包含多达18个生物学条件、8个生物学重复且每个样本有1亿(M)条 reads。作为一个总体趋势,我们得出结论,除了低表达基因外,重复样本数量对DE分析功效的影响比文库大小更大,对于低表达基因,这两个参数似乎具有相同的影响。我们的研究还为旨在改进实验设计的从业者提供了新的见解。例如,通过分析DE分析的敏感性和特异性,我们表明控制错误发现率(FDR)的最佳阈值约为2,其中r是重复样本数量。此外,我们表明所有三个研究的R包: 、 和 对误报率(FPR)的控制相当良好。我们还分析了重复样本数量和文库大小对基因本体(GO)富集分析的影响。有趣的是,我们得出结论,重复样本数量和文库大小的增加往往分别提高GO分析的敏感性和特异性。最后,我们建议RNA测序从业者生成一个试点数据集,以严格分析其实验设计的功效,或者使用一个公共数据集,该数据集应与他们将获得的数据集相似。对于从事番茄研究的人员,基于荟萃分析,我们建议每个条件至少有四个生物学重复且每个样本有20M条reads,以便几乎肯定能获得约1000个DE基因(如果存在的话)。