Schurch Nicholas J, Schofield Pietá, Gierliński Marek, Cole Christian, Sherstnev Alexander, Singh Vijender, Wrobel Nicola, Gharbi Karim, Simpson Gordon G, Owen-Hughes Tom, Blaxter Mark, Barton Geoffrey J
Division of Computational Biology, College of Life Sciences, University of Dundee, Dundee DD1 5EH, United Kingdom.
Division of Computational Biology, College of Life Sciences, University of Dundee, Dundee DD1 5EH, United Kingdom Division of Gene Regulation and Expression, College of Life Sciences, University of Dundee, Dundee DD1 5EH, United Kingdom.
RNA. 2016 Jun;22(6):839-51. doi: 10.1261/rna.053959.115. Epub 2016 Mar 28.
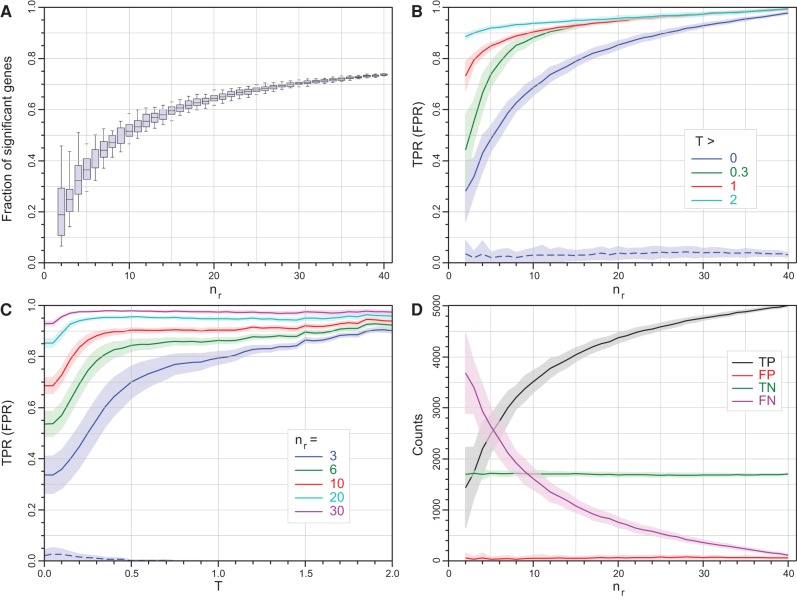
RNA-seq is now the technology of choice for genome-wide differential gene expression experiments, but it is not clear how many biological replicates are needed to ensure valid biological interpretation of the results or which statistical tools are best for analyzing the data. An RNA-seq experiment with 48 biological replicates in each of two conditions was performed to answer these questions and provide guidelines for experimental design. With three biological replicates, nine of the 11 tools evaluated found only 20%-40% of the significantly differentially expressed (SDE) genes identified with the full set of 42 clean replicates. This rises to >85% for the subset of SDE genes changing in expression by more than fourfold. To achieve >85% for all SDE genes regardless of fold change requires more than 20 biological replicates. The same nine tools successfully control their false discovery rate at ≲5% for all numbers of replicates, while the remaining two tools fail to control their FDR adequately, particularly for low numbers of replicates. For future RNA-seq experiments, these results suggest that at least six biological replicates should be used, rising to at least 12 when it is important to identify SDE genes for all fold changes. If fewer than 12 replicates are used, a superior combination of true positive and false positive performances makes edgeR and DESeq2 the leading tools. For higher replicate numbers, minimizing false positives is more important and DESeq marginally outperforms the other tools.
RNA测序如今是全基因组差异基因表达实验的首选技术,但尚不清楚需要多少生物学重复才能确保对结果进行有效的生物学解读,也不清楚哪种统计工具最适合分析数据。为回答这些问题并为实验设计提供指导方针,进行了一项RNA测序实验,在两种条件下每种条件均设置48个生物学重复。对于11种评估工具中的9种而言,若只有3个生物学重复,那么它们所发现的显著差异表达(SDE)基因仅占利用全部42个有效重复鉴定出的SDE基因的20%-40%。对于表达变化超过四倍的SDE基因子集,这一比例升至>85%。要使所有SDE基因无论其倍数变化如何都能达到>85%的比例,则需要超过20个生物学重复。对于所有重复数量,同样的9种工具都能成功将其错误发现率控制在≲5%,而其余两种工具无法充分控制其错误发现率,尤其是在重复数量较少时。对于未来的RNA测序实验,这些结果表明应至少使用6个生物学重复,若要识别所有倍数变化的SDE基因,这一数量应至少增至12个。如果使用的重复数量少于12个,由于真阳性和假阳性表现的出色组合,edgeR和DESeq2成为领先工具。对于更多的重复数量,将假阳性降至最低更为重要,DESeq略优于其他工具。