Department of Cardiology, Chinese PLA General Hospital, Beijing, China.
Department of Chemical and Environmental Engineering, University of California, Riverside, CA 92521 USA.
Redox Biol. 2018 Jun;16:157-168. doi: 10.1016/j.redox.2018.02.019. Epub 2018 Mar 1.
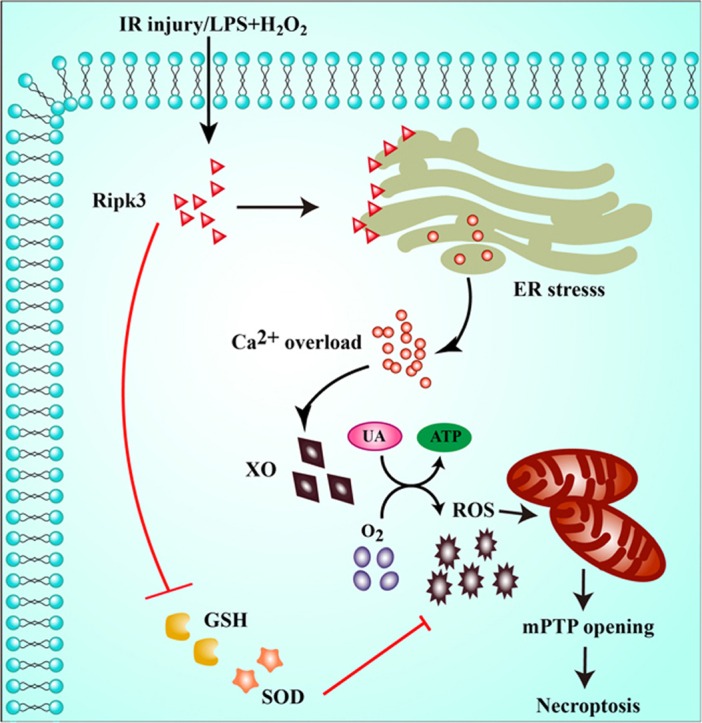
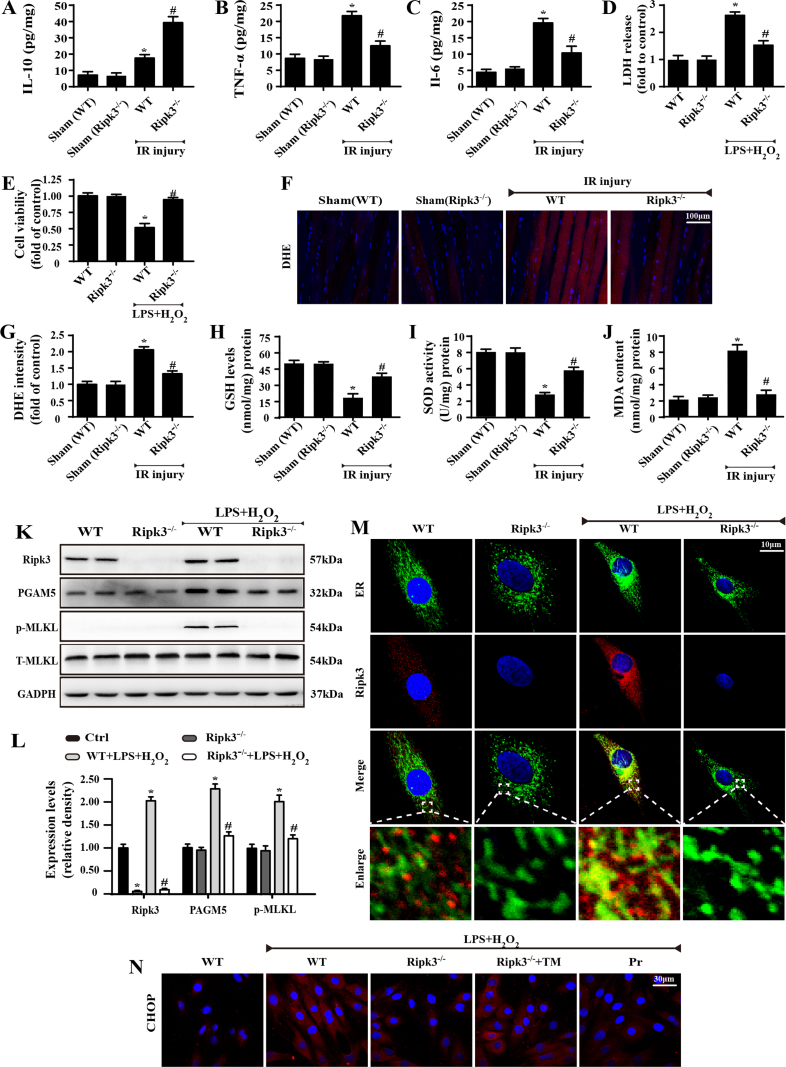
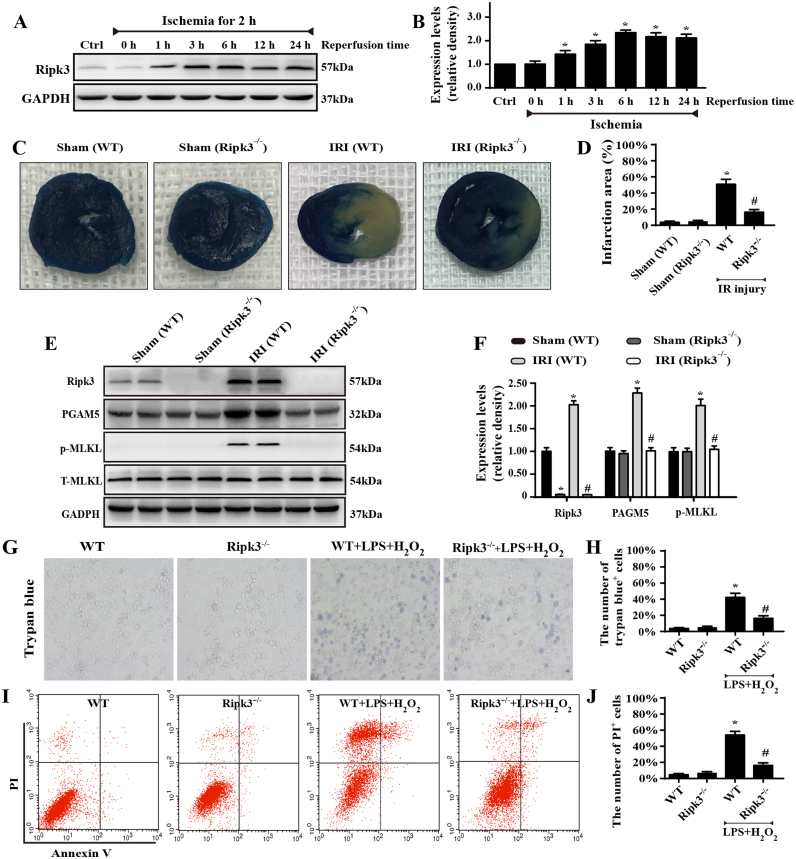
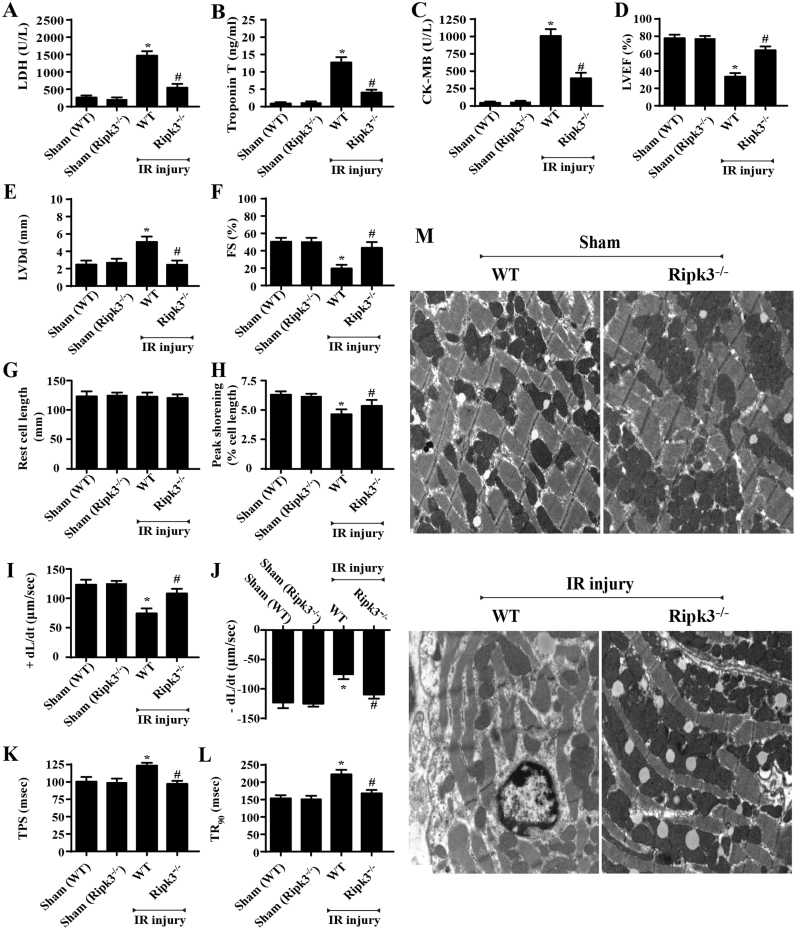
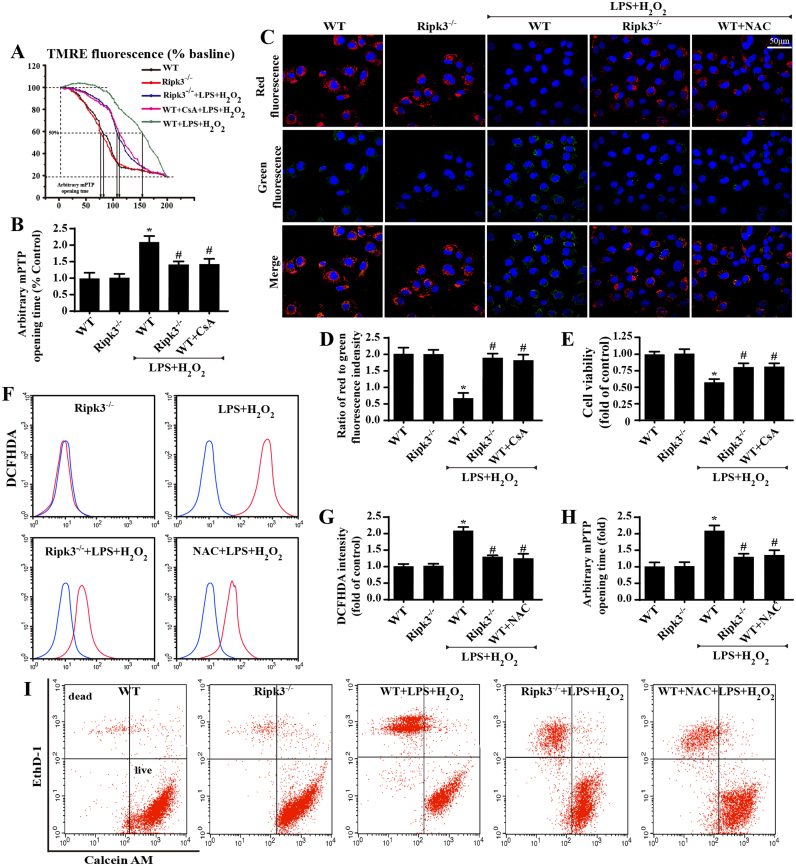
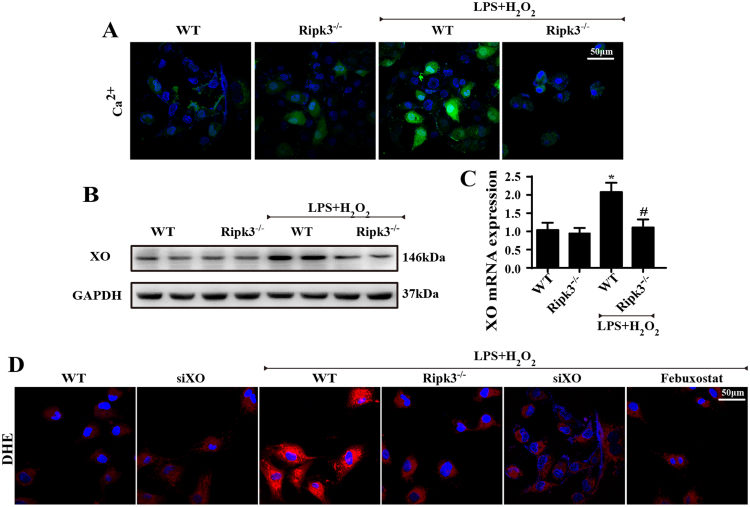
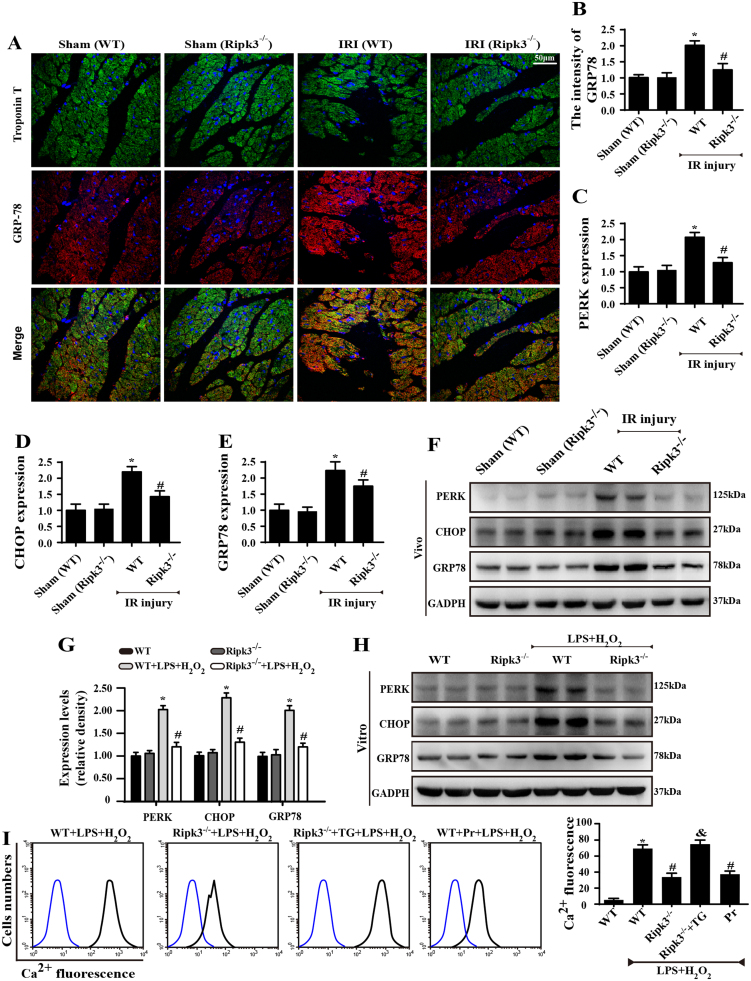
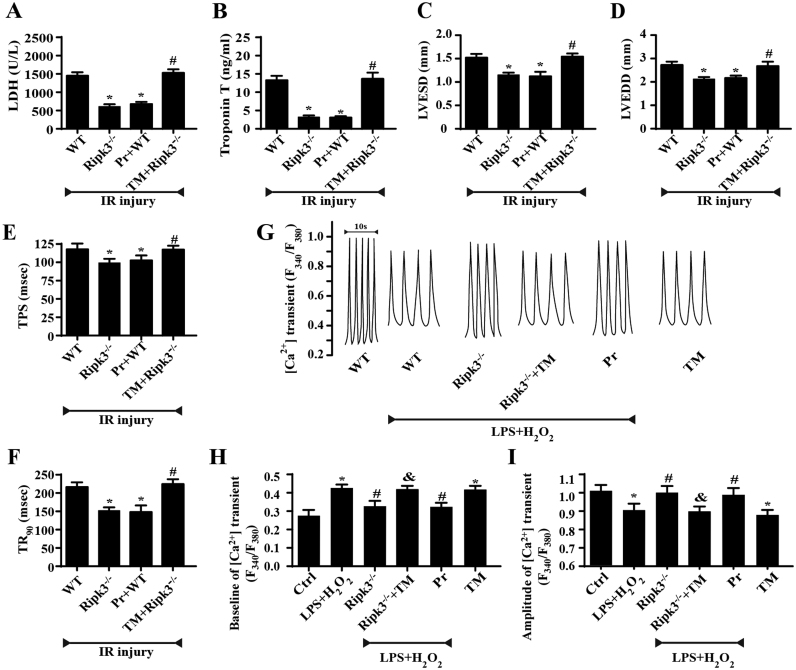
Receptor-interacting protein 3 (Ripk3)-mediated necroptosis contributes to cardiac ischaemia-reperfusion (IR) injury through poorly defined mechanisms. Our results demonstrated that Ripk3 was strongly upregulated in murine hearts subjected to IR injury and cardiomyocytes treated with LPS and HO. The higher level of Ripk3 was positively correlated to the infarction area expansion, cardiac dysfunction and augmented cardiomyocytes necroptosis. Function study further illustrated that upregulated Ripk3 evoked the endoplasmic reticulum (ER) stress, which was accompanied with an increase in intracellular Ca level ([Ca]c) and xanthine oxidase (XO) expression. Activated XO raised cellular reactive oxygen species (ROS) that mediated the mitochondrial permeability transition pore (mPTP) opening and cardiomyocytes necroptosis. By comparison, genetic ablation of Ripk3 abrogated the ER stress and thus blocked the [Ca]c overload-XO-ROS-mPTP pathways, favouring a pro-survival state that ultimately resulted in the inhibition of cardiomyocytes necroptosis in the setting of cardiac IR injury. In summary, the present study helps to elucidate how necroptosis is mediated by ER stress, via the calcium overload /XO/ROS/mPTP opening axis.
受体相互作用蛋白 3(Ripk3)介导的坏死性凋亡通过未明确的机制导致心脏缺血再灌注(IR)损伤。我们的结果表明,Ripk3 在经历 IR 损伤的小鼠心脏和接受 LPS 和 HO 处理的心肌细胞中强烈上调。Ripk3 的高水平与梗死面积扩大、心脏功能障碍和增强的心肌细胞坏死性凋亡呈正相关。功能研究进一步表明,上调的 Ripk3 引发内质网(ER)应激,这伴随着细胞内 Ca 水平 ([Ca]c) 和黄嘌呤氧化酶 (XO) 表达的增加。激活的 XO 增加细胞活性氧 (ROS),介导线粒体通透性转换孔 (mPTP) 开放和心肌细胞坏死性凋亡。相比之下,Ripk3 的基因缺失消除了 ER 应激,从而阻断了 [Ca]c 过载-XO-ROS-mPTP 途径,有利于生存状态,最终导致心脏 IR 损伤时心肌细胞坏死性凋亡的抑制。总之,本研究有助于阐明坏死性凋亡如何通过内质网应激介导,通过钙超载/XO/ROS/mPTP 开放轴。