Center for Computational Biology and Department of Molecular Biosciences, The University of Kansas, Lawrence, Kansas, 66047, USA.
BMC Bioinformatics. 2018 Mar 5;19(1):84. doi: 10.1186/s12859-018-2079-4.
Structural modeling of protein-protein interactions produces a large number of putative configurations of the protein complexes. Identification of the near-native models among them is a serious challenge. Publicly available results of biomedical research may provide constraints on the binding mode, which can be essential for the docking. Our text-mining (TM) tool, which extracts binding site residues from the PubMed abstracts, was successfully applied to protein docking (Badal et al., PLoS Comput Biol, 2015; 11: e1004630). Still, many extracted residues were not relevant to the docking.
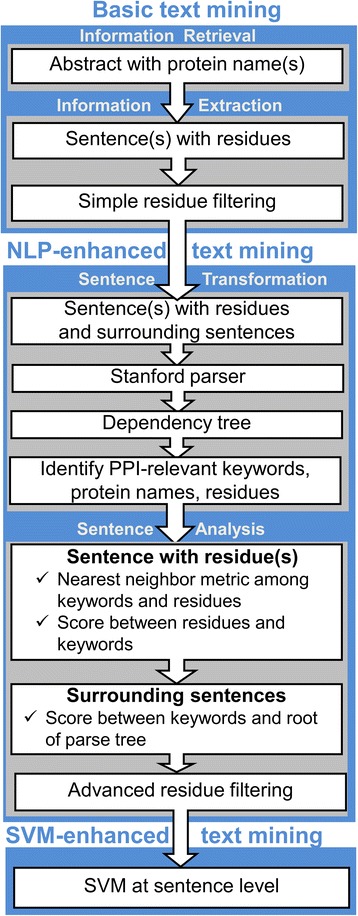
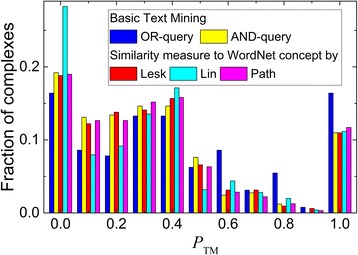
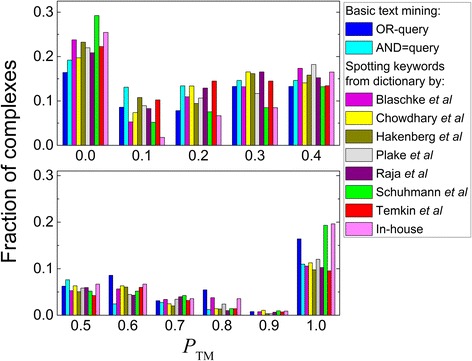
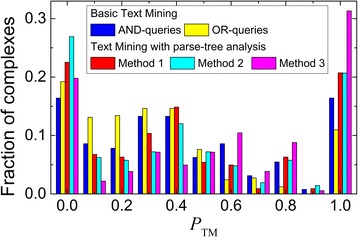
We present an extension of the TM tool, which utilizes natural language processing (NLP) for analyzing the context of the residue occurrence. The procedure was tested using generic and specialized dictionaries. The results showed that the keyword dictionaries designed for identification of protein interactions are not adequate for the TM prediction of the binding mode. However, our dictionary designed to distinguish keywords relevant to the protein binding sites led to considerable improvement in the TM performance. We investigated the utility of several methods of context analysis, based on dissection of the sentence parse trees. The machine learning-based NLP filtered the pool of the mined residues significantly more efficiently than the rule-based NLP. Constraints generated by NLP were tested in docking of unbound proteins from the DOCKGROUND X-ray benchmark set 4. The output of the global low-resolution docking scan was post-processed, separately, by constraints from the basic TM, constraints re-ranked by NLP, and the reference constraints. The quality of a match was assessed by the interface root-mean-square deviation. The results showed significant improvement of the docking output when using the constraints generated by the advanced TM with NLP.
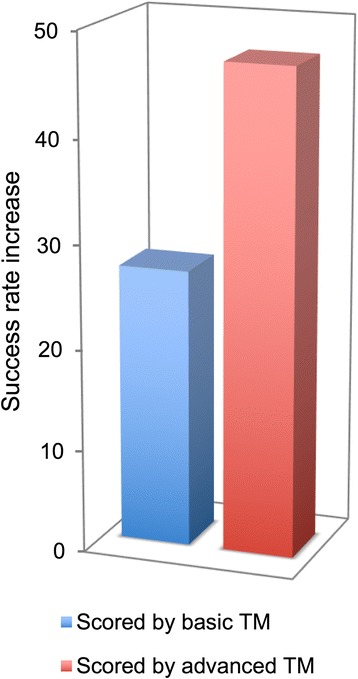
The basic TM procedure for extracting protein-protein binding site residues from the PubMed abstracts was significantly advanced by the deep parsing (NLP techniques for contextual analysis) in purging of the initial pool of the extracted residues. Benchmarking showed a substantial increase of the docking success rate based on the constraints generated by the advanced TM with NLP.
蛋白质-蛋白质相互作用的结构建模会产生大量蛋白质复合物的假定构象。在这些构象中识别接近天然的模型是一个严峻的挑战。公共生物医学研究结果可提供结合模式的约束条件,这对于对接至关重要。我们的从 PubMed 摘要中提取结合位点残基的文本挖掘(TM)工具已成功应用于蛋白质对接(Badal 等人, PLoS Comput Biol,2015;11:e1004630)。尽管如此,许多提取的残基与对接无关。
我们提出了 TM 工具的扩展,该工具利用自然语言处理(NLP)来分析残基出现的上下文。该程序使用通用和专用词典进行了测试。结果表明,用于识别蛋白质相互作用的关键字词典对于 TM 预测结合模式是不够的。然而,我们设计的用于区分与蛋白质结合位点相关的关键字的词典导致 TM 性能的显著提高。我们研究了基于句子解析树切割的几种上下文分析方法的实用性。基于机器学习的 NLP 比基于规则的 NLP 更有效地过滤挖掘出的残基池。在对接 DOCKGROUND X 射线基准集 4 中的未结合蛋白质时,对 NLP 生成的约束进行了测试。全局低分辨率对接扫描的输出分别通过基本 TM 的约束、由 NLP 重新排序的约束和参考约束进行后处理。通过接口均方根偏差评估匹配的质量。结果表明,当使用具有 NLP 的高级 TM 生成的约束时,对接输出得到了显著改善。
通过深度解析(用于上下文分析的 NLP 技术)对从 PubMed 摘要中提取蛋白质-蛋白质结合位点残基的基本 TM 过程进行了显著改进,从而清除了初始提取残基池。基准测试表明,基于具有 NLP 的高级 TM 生成的约束条件,对接成功率有了实质性提高。