Henan Center for Disease Control and Prevention, Zhengzhou, Henan, China.
National Institute of Parasitic Diseases, Chinese Center for Disease Control and Prevention, WHO Collaborating Centre for Malaria, Schistosomiasis and Filariasis, Key Laboratory of Parasite and Vector Biology, Ministry of Health, Shanghai, China.
Malar J. 2018 Mar 5;17(1):103. doi: 10.1186/s12936-018-2237-1.
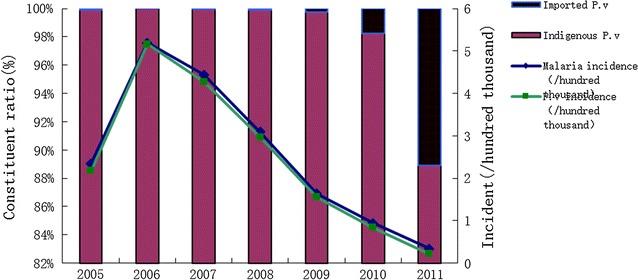
Plasmodium vivax malaria has historically been a major source of disease in Henan, China. In the 1970s, the morbidity of malaria was highest in the country. With support from the government and the efforts of healthcare personnel, the reported malaria cases have declined dramatically and a national elimination programme was launched in 2010. To achieve the goal, it is essential to study the diversity of autochthonous malaria and transmission of Plasmodium parasites, which will provide baseline data for disease control and management.
Thirty-two P. vivax isolates from Henan province were collected from 2008 to 2011, and circumsporozoite protein (csp) genes were analysed to estimate the genetic diversity of this parasite.
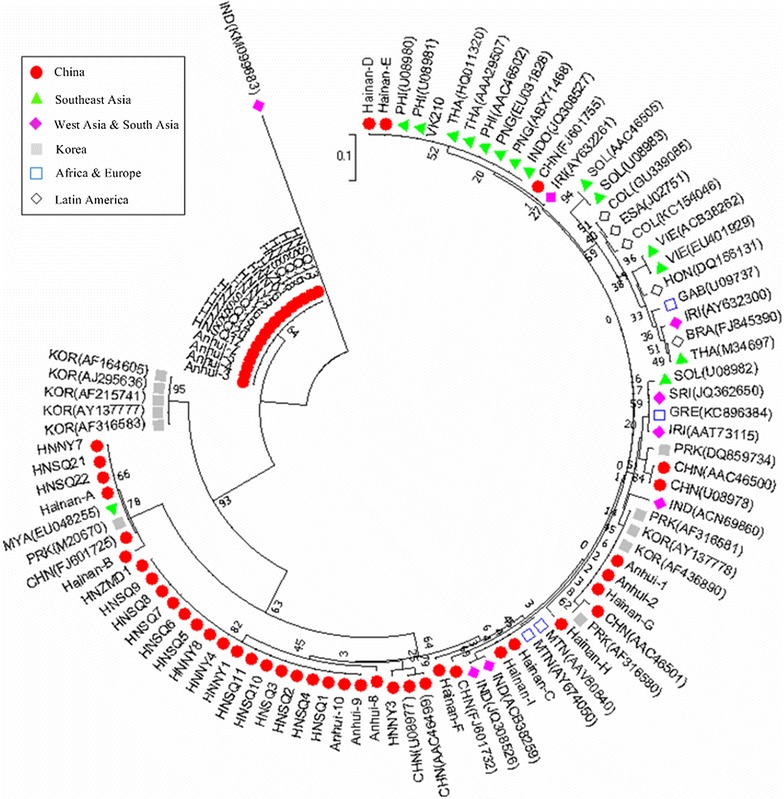
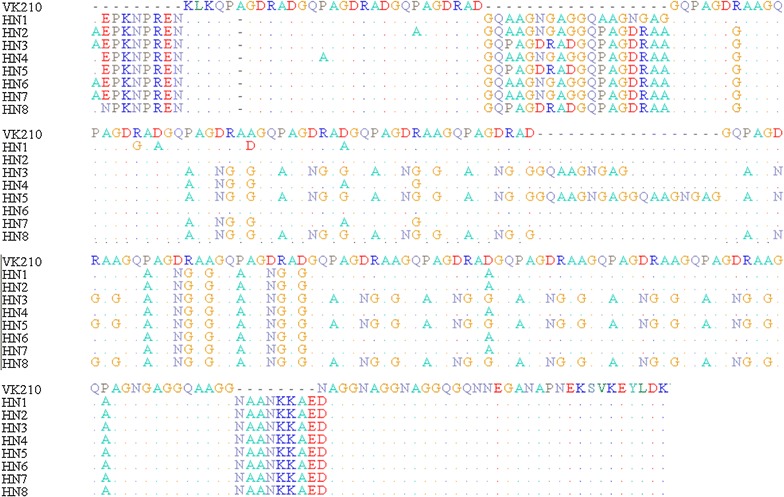
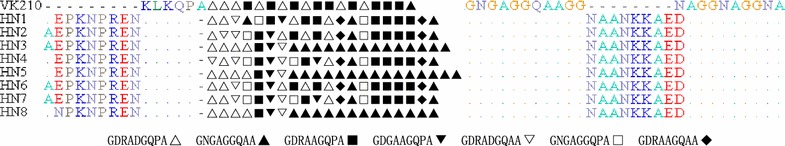
The assessment of csp sequences indicated that all the isolates were the VK210 type, however, none of them was identical to the VK210 strain. The sequences displayed variations in the central region, and eight sub-types were observed. Among the sub-types, HN7 was the most prevalent (37.5%), followed by HN3 (34.4%). A total of 653 repeat units were discovered in 32 Henan isolates. Nucleotide sequences were grouped in 13 unique repeat nucleotide sequence allotypes that coded for 7 different repeated amino acid allotypes. B (GNGAGGQAA) and D (GDRAAGQPA) were more frequent based on the results; they represented 53.9% (352/653) of the total. In comparison to the basic repeat units of VK210, more than 75% of the central repeat units had at least one non-synonymous nucleotide change.
Recent P. vivax populations in Henan province showed some degree of genetic diversity in csp, with 8 sub-types among 32 samples. Meantime, the results also suggested its relative conserved parasite populations. This could provide interesting baseline data that allow identifying whether potential new cases differ from the parasites already circulating in the area.
间日疟原虫疟疾曾是中国河南的主要疾病来源。20 世纪 70 年代,中国的疟疾发病率最高。在政府的支持和医务人员的努力下,报告的疟疾病例大幅下降,并于 2010 年启动了国家消除规划。为实现这一目标,必须研究当地疟原虫的多样性和疟原虫寄生虫的传播,这将为疾病控制和管理提供基线数据。
2008 年至 2011 年从河南省采集了 32 株间日疟原虫分离株,分析环子孢子蛋白(csp)基因,以估计该寄生虫的遗传多样性。
对 csp 序列的评估表明,所有分离株均为 VK210 型,但均与 VK210 株不同。序列在中央区域显示出变异,观察到 8 种亚型。在亚型中,HN7 最为常见(37.5%),其次是 HN3(34.4%)。在 32 株河南分离株中发现了 653 个重复单位。核苷酸序列分为 13 个独特的重复核苷酸序列等位基因,编码 7 种不同的重复氨基酸等位基因。根据结果,B(GNGAGGQAA)和 D(GDRAAGQPA)更为常见;它们占总数的 53.9%(352/653)。与 VK210 的基本重复单位相比,中央重复单位中超过 75%的单位至少有一个非同义核苷酸变化。
河南省近期间日疟原虫种群在 csp 中表现出一定程度的遗传多样性,32 个样本中有 8 个亚型。同时,结果还表明其寄生虫种群相对保守。这可以提供有趣的基线数据,以确定潜在的新病例是否与该地区已流行的寄生虫不同。