Butera Diego, Passam Freda, Ju Lining, Cook Kristina M, Woon Heng, Aponte-Santamaría Camilo, Gardiner Elizabeth, Davis Amanda K, Murphy Deirdre A, Bronowska Agnieszka, Luken Brenda M, Baldauf Carsten, Jackson Shaun, Andrews Robert, Gräter Frauke, Hogg Philip J
The Centenary Institute, Newtown, New South Wales, Australia.
St George Clinical School, Kogarah, New South Wales, Australia.
Sci Adv. 2018 Feb 28;4(2):eaaq1477. doi: 10.1126/sciadv.aaq1477. eCollection 2018 Feb.
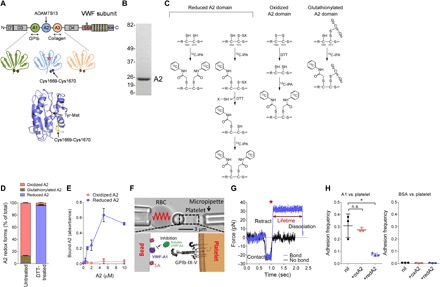
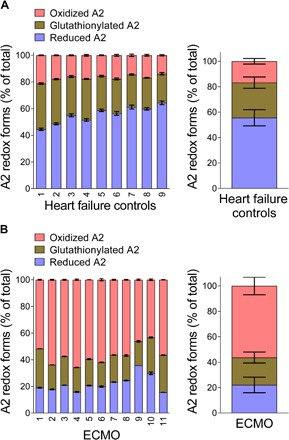
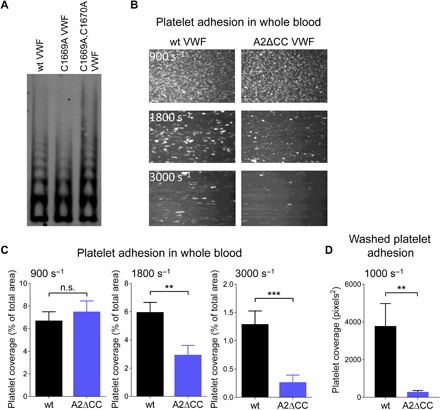
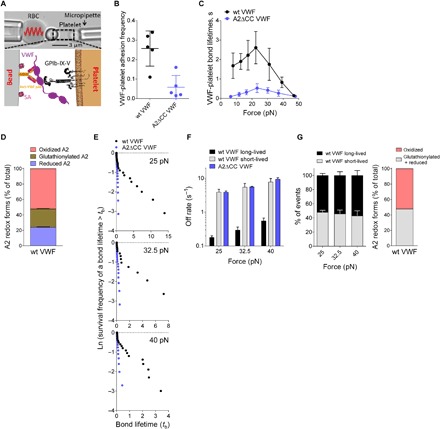
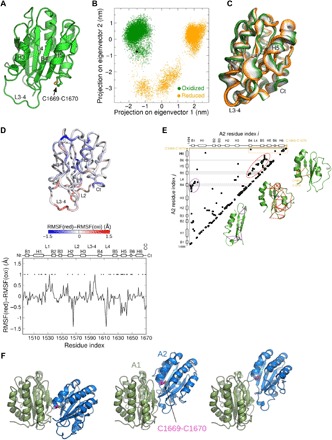
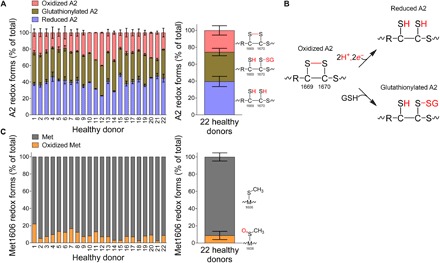
Force-dependent binding of platelet glycoprotein Ib (GPIb) receptors to plasma von Willebrand factor (VWF) plays a key role in hemostasis and thrombosis. Previous studies have suggested that VWF activation requires force-induced exposure of the GPIb binding site in the A1 domain that is autoinhibited by the neighboring A2 domain. However, the biochemical basis of this "mechanopresentation" remains elusive. From a combination of protein chemical, biophysical, and functional studies, we find that the autoinhibition is controlled by the redox state of an unusual disulfide bond near the carboxyl terminus of the A2 domain that links adjacent cysteine residues to form an eight-membered ring. Only when the bond is cleaved does the A2 domain bind to the A1 domain and block platelet GPIb binding. Molecular dynamics simulations indicate that cleavage of the disulfide bond modifies the structure and molecular stresses of the A2 domain in a long-range allosteric manner, which provides a structural explanation for redox control of the autoinhibition. Significantly, the A2 disulfide bond is cleaved in ~75% of VWF subunits in healthy human donor plasma but in just ~25% of plasma VWF subunits from heart failure patients who have received extracorporeal membrane oxygenation support. This suggests that the majority of plasma VWF binding sites for platelet GPIb are autoinhibited in healthy donors but are mostly available in heart failure patients. These findings demonstrate that a disulfide bond switch regulates mechanopresentation of VWF.
血小板糖蛋白Ib(GPIb)受体与血浆血管性血友病因子(VWF)的力依赖性结合在止血和血栓形成中起关键作用。先前的研究表明,VWF激活需要在A1结构域中由相邻的A2结构域自动抑制的GPIb结合位点通过力诱导暴露。然而,这种“机械呈递”的生化基础仍然难以捉摸。通过蛋白质化学、生物物理和功能研究的结合,我们发现自动抑制由A2结构域羧基末端附近一个不寻常的二硫键的氧化还原状态控制,该二硫键连接相邻的半胱氨酸残基形成一个八元环。只有当该键断裂时,A2结构域才会与A1结构域结合并阻断血小板GPIb结合。分子动力学模拟表明,二硫键的断裂以长程变构方式改变了A2结构域的结构和分子应力,这为自动抑制的氧化还原控制提供了结构解释。值得注意的是,在健康人类供体血浆中,约75%的VWF亚基中的A2二硫键被切割,但在接受体外膜肺氧合支持的心力衰竭患者的血浆VWF亚基中,只有约25%被切割。这表明,在健康供体中,血小板GPIb的大多数血浆VWF结合位点被自动抑制,但在心力衰竭患者中大多可用。这些发现表明,二硫键开关调节VWF的机械呈递。