Department of Medical Statistics, University Medical Center Göttingen, 37073, Göttingen, Germany.
Division Applied Bioinformatics, German Cancer Research Center (DKFZ) and National Center for Tumor Diseases (NCT), 69120, Heidelberg, Germany.
Genome Med. 2018 Mar 15;10(1):18. doi: 10.1186/s13073-018-0529-2.
A comprehensive understanding of cancer has been furthered with technological improvements and decreasing costs of next-generation sequencing (NGS). However, the complexity of interpreting genomic data is hindering the implementation of high-throughput technologies in the clinical context: increasing evidence on gene-drug interactions complicates the task of assigning clinical significance to genomic variants.
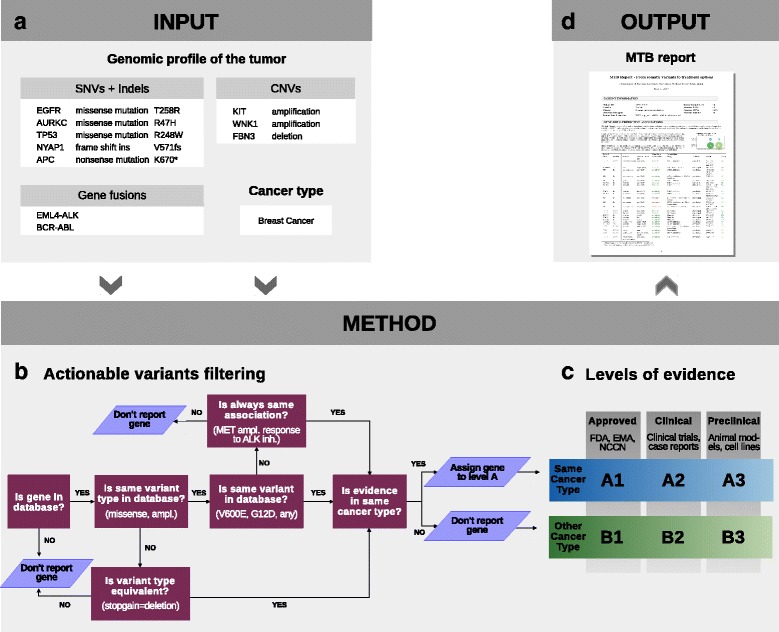
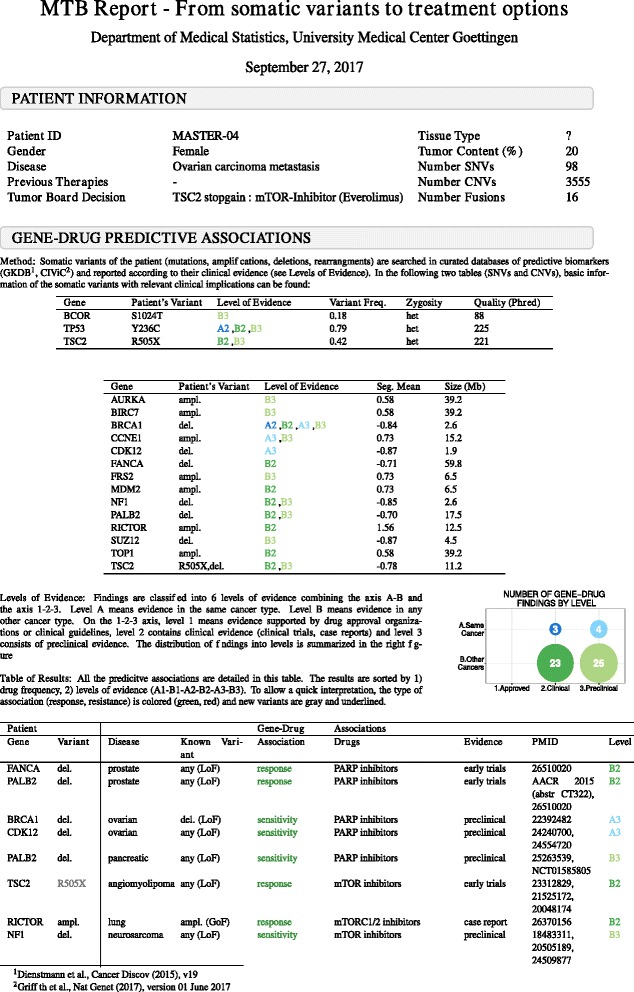
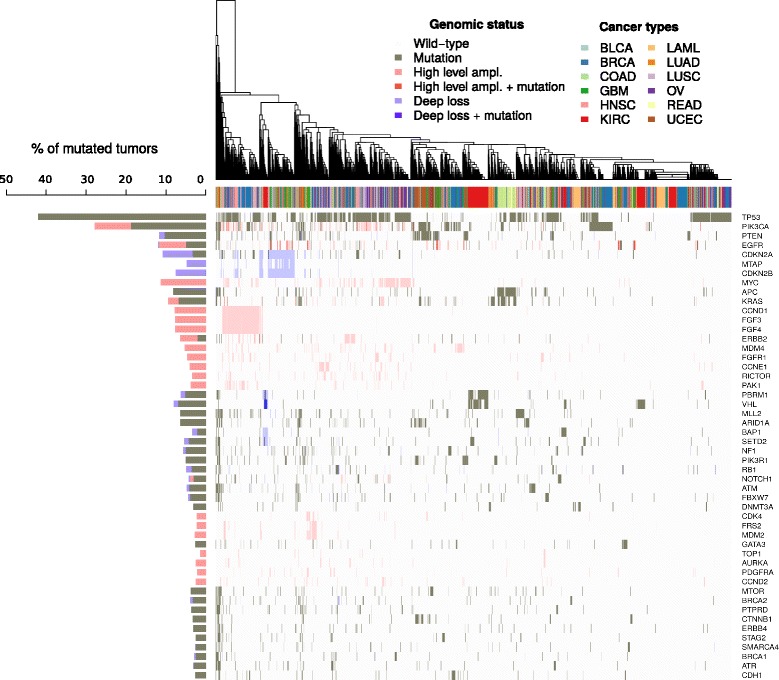
Here we present a method that automatically matches patient-specific genomic alterations to treatment options. The method relies entirely on public knowledge of somatic variants with predictive evidence on drug response. The output report is aimed at supporting clinicians in the task of finding the clinical meaning of genomic variants. We applied the method to 1) The Cancer Genome Atlas (TCGA) and Genomics Evidence Neoplasia Information Exchange (GENIE) cohorts and 2) 11 patients from the NCT MASTER trial whose treatment discussions included information on their genomic profiles.
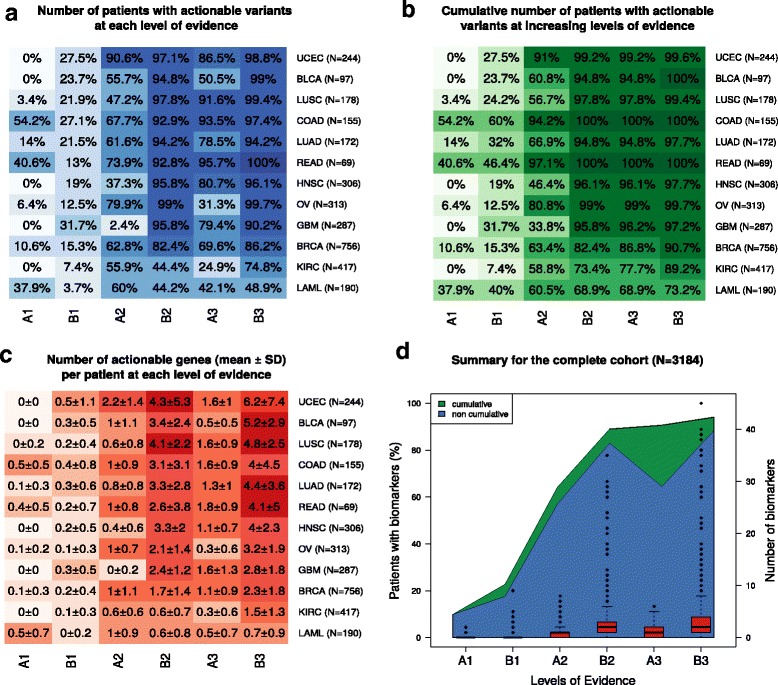
Our reporting strategy showed a substantial number of patients with actionable variants in the analyses of TCGA and GENIE samples. Notably, it was able to reproduce experts' treatment suggestions in a retrospective study of 11 patients from the NCT MASTER trial. Our results establish a proof of concept for comprehensive, evidence-based reports as a supporting tool for discussing treatment options in tumor boards.
We believe that a standardized method to report actionable somatic variants will smooth the incorporation of NGS in the clinical context. We anticipate that tools like the one we present here will become essential in summarizing for clinicians the growing evidence in the field of precision medicine. The R code of the presented method is provided in Additional file 6 and available at https://github.com/jperera-bel/MTB-Report .
随着下一代测序(NGS)技术的改进和成本的降低,对癌症的全面认识得到了进一步提高。然而,基因组数据解释的复杂性阻碍了高通量技术在临床环境中的应用:越来越多的基因-药物相互作用的证据使得将基因组变异赋予临床意义的任务变得复杂。
本文提出了一种自动将患者特异性基因组改变与治疗选择相匹配的方法。该方法完全依赖于具有药物反应预测证据的体细胞变异的公共知识。输出报告旨在帮助临床医生找到基因组变异的临床意义。我们将该方法应用于 1)癌症基因组图谱(TCGA)和基因组证据肿瘤信息交换(GENIE)队列,以及 2)来自 NCT MASTER 试验的 11 名患者,这些患者的治疗讨论包括其基因组谱的信息。
我们的报告策略在 TCGA 和 GENIE 样本的分析中显示了大量具有可操作变异的患者。值得注意的是,它能够在对 NCT MASTER 试验的 11 名患者的回顾性研究中重现专家的治疗建议。我们的结果为全面的、基于证据的报告提供了一个概念验证,作为在肿瘤委员会讨论治疗选择的支持工具。
我们相信,报告可操作体细胞变异的标准化方法将使 NGS 在临床环境中的应用更加顺畅。我们预计,像我们在这里提出的这样的工具将成为总结精准医学领域不断增长的证据的重要工具。本文所提出方法的 R 代码在附加文件 6 中提供,并可在 https://github.com/jperera-bel/MTB-Report 上获得。