Li Yimeng, Hu Xiaolong, Yang Shuang, Zhou Juntong, Qi Lei, Sun Xiaoning, Fan Mengyuan, Xu Shanghua, Cha Muha, Zhang Meishan, Lin Shaobi, Liu Shuqiang, Hu Defu
College of Nature Conservation, Beijing Forestry University, Beijing, China.
College of Animal Science and Technology, Jiangxi Agricultural University, Nanchang, China.
Front Microbiol. 2018 Mar 2;9:300. doi: 10.3389/fmicb.2018.00300. eCollection 2018.
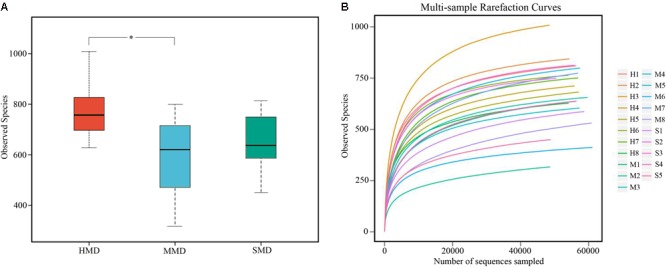
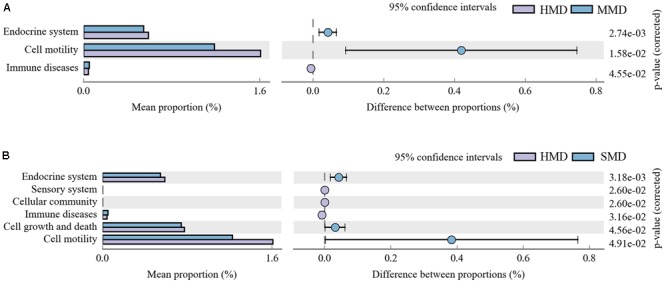
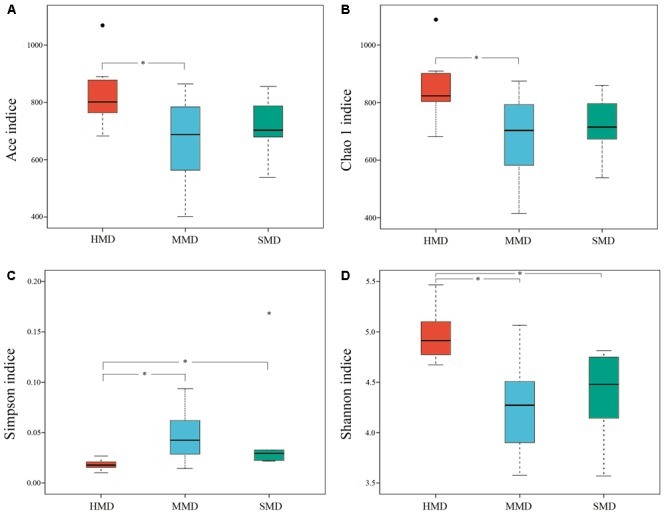
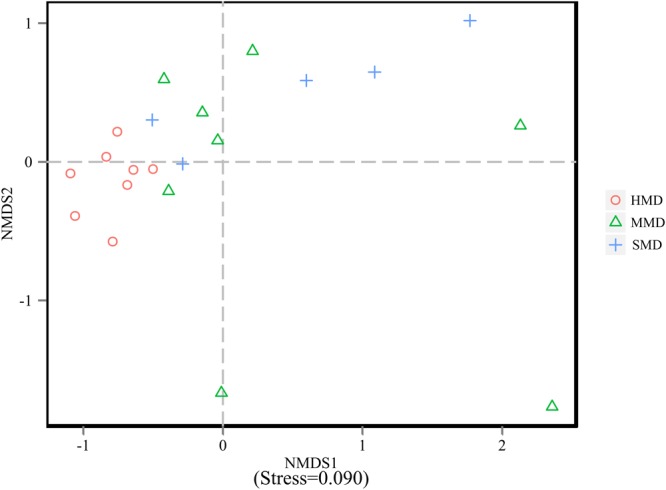
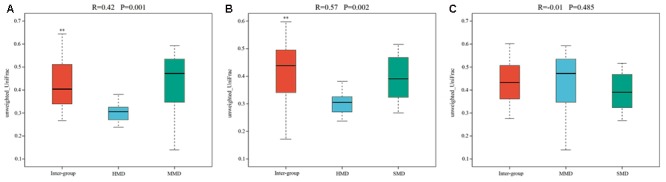
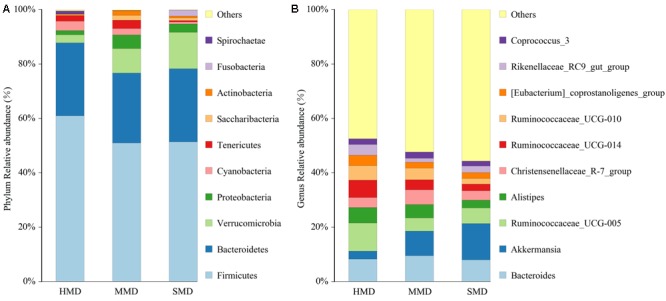
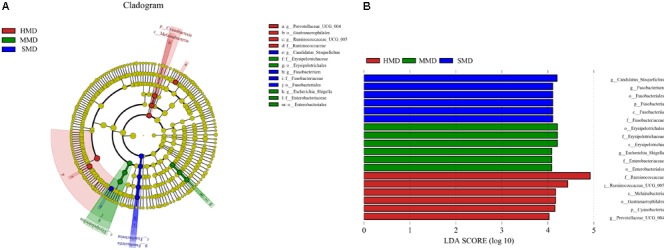
Diarrhea constitutes one of the most common diseases affecting the survival of captive musk deer and is usually caused by an imbalance in intestinal microbiota. Currently, research regarding the structure and function of intestinal microbiota in diarrheic musk deer is lacking. Therefore, in the present study, high-throughput 16S-rRNA gene sequencing was used to analyze the intestinal microbiota in feces of healthy captive musk deer (HMD) ( = 8) and musk deer with mild (MMD) ( = 8), and severe ( = 5) (SMD) diarrhea to compare the difference in intestinal microbiota of musk deer under various physiological conditions. The results showed that the diversity of HMD fecal microbiota was significantly higher than that of the two diarrhea samples. β Diversity results indicated that there were extremely significant differences in bacterial communities between the HMD sample and the MMD and SMD samples. However, no significant difference was found between the two diarrhea samples. LefSe analysis showed that the degree of intestinal physiological dysfunction in musk deer was correlated with the types of major pathogens. The main pathogen in the MMD group is , whereas is the main pathogen in the SMD group. PICRUSt functional profile prediction indicated that the intestinal microbiota disorder could also lead to changes in the abundance of genes in metabolic pathways of the immune system. Altogether, this study provides a theoretical basis for the exploration of treatments for diarrhea in captive musk deer, which is of considerable significance to the implementation of the musk deer release into the wild program.
腹泻是影响圈养林麝生存的最常见疾病之一,通常由肠道微生物群失衡引起。目前,关于腹泻林麝肠道微生物群的结构和功能的研究尚缺。因此,在本研究中,采用高通量16S - rRNA基因测序分析健康圈养林麝(HMD)(n = 8)、轻度腹泻林麝(MMD)(n = 8)和重度腹泻林麝(SMD)(n = 5)粪便中的肠道微生物群,以比较不同生理条件下林麝肠道微生物群的差异。结果表明,HMD粪便微生物群的多样性显著高于两个腹泻样本。β多样性结果表明,HMD样本与MMD和SMD样本之间的细菌群落存在极显著差异。然而,两个腹泻样本之间未发现显著差异。LefSe分析表明,林麝肠道生理功能障碍程度与主要病原体类型相关。MMD组的主要病原体是 ,而SMD组的主要病原体是 。PICRUSt功能谱预测表明,肠道微生物群紊乱也可能导致免疫系统代谢途径中基因丰度的变化。总之,本研究为探索圈养林麝腹泻的治疗方法提供了理论依据,对实施林麝放归野外计划具有重要意义。