a Department of Enteric Infections, The Hospital for Tropical Diseases, Wellcome Trust Major Overseas Programme , Oxford University Clinical Research Unit , Ho Chi Minh City , Vietnam.
b The Genome Institute of Singapore, GIS Efficient Rapid Microbial Sequencing (GERMS) , Singapore.
Gut Microbes. 2018 Jan 2;9(1):38-54. doi: 10.1080/19490976.2017.1361093. Epub 2017 Aug 24.
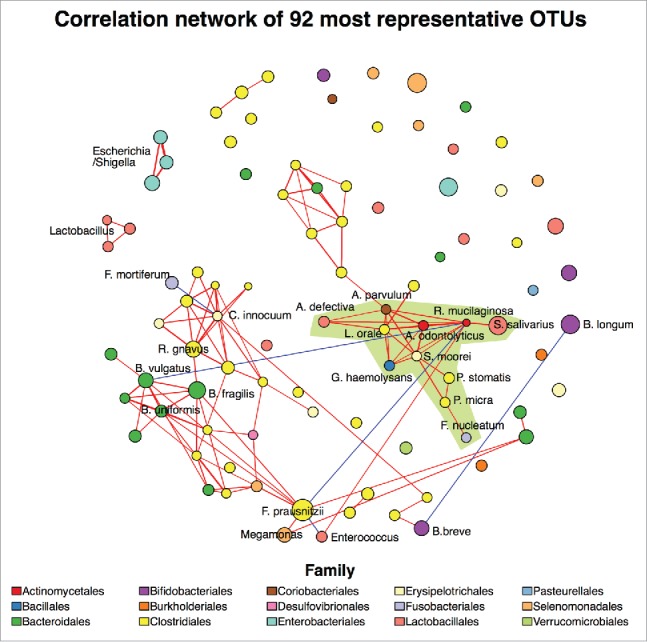
Diarrheal diseases remain the second most common cause of mortality in young children in developing countries. Efforts have been made to explore the impact of diarrhea on bacterial communities in the human gut, but a thorough understanding has been impeded by inadequate resolution in bacterial identification and the examination of only few etiological agents. Here, by profiling an extended region of the 16S rRNA gene in the fecal microbiome, we aimed to elucidate the nature of gut microbiome perturbations during the early phase of infectious diarrhea caused by various etiological agents in Vietnamese children. Fecal samples from 145 diarrheal cases with a confirmed infectious etiology before antimicrobial therapy and 54 control subjects were analyzed. We found that the diarrheal fecal microbiota could be robustly categorized into 4 microbial configurations that either generally resembled or were highly divergent from a healthy state. Factors such as age, nutritional status, breastfeeding, and the etiology of the infection were significantly associated with these microbial community structures. We observed a consistent elevation of Fusobacterium mortiferum, Escherichia, and oral microorganisms in all diarrheal fecal microbiome configurations, proposing similar mechanistic interactions, even in the absence of global dysbiosis. We additionally found that Bifidobacterium pseudocatenulatum was significantly depleted during dysenteric diarrhea regardless of the etiological agent, suggesting that further investigations into the use of this species as a dysentery-orientated probiotic therapy are warranted. Our findings contribute to the understanding of the complex influence of infectious diarrhea on gut microbiome and identify new opportunities for therapeutic interventions.
腹泻病仍然是发展中国家幼儿死亡的第二大主要原因。人们一直在努力探索腹泻对人类肠道细菌群落的影响,但由于细菌鉴定的分辨率不足以及仅检查了少数几种病因,因此对其的全面了解受到了阻碍。在这里,我们通过对粪便微生物组中 16S rRNA 基因的扩展区域进行分析,旨在阐明越南儿童因各种病因引起的感染性腹泻早期阶段肠道微生物组紊乱的性质。在进行抗生素治疗之前,对 145 例经确认具有传染性病因的腹泻病例和 54 例对照的粪便样本进行了分析。我们发现,腹泻粪便微生物组可以分为 4 种微生物构型,这些构型要么与健康状态相似,要么与健康状态高度不同。年龄、营养状况、母乳喂养和感染的病因等因素与这些微生物群落结构显著相关。我们观察到所有腹泻粪便微生物组构型中均一致存在梭菌属和大肠杆菌以及口腔微生物的升高,这表明即使没有全球菌群失调,也存在类似的机制相互作用。我们还发现,无论病因如何,双歧杆菌假链状均在痢疾性腹泻中明显减少,这表明有必要进一步研究将该物种用作痢疾定向益生菌治疗的方法。我们的研究结果有助于理解感染性腹泻对肠道微生物组的复杂影响,并为治疗干预提供了新的机会。