Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, Canada.
Department of Medical Biophysics, University of Toronto, Toronto, Ontario, Canada.
PLoS Comput Biol. 2018 Mar 28;14(3):e1006080. doi: 10.1371/journal.pcbi.1006080. eCollection 2018 Mar.
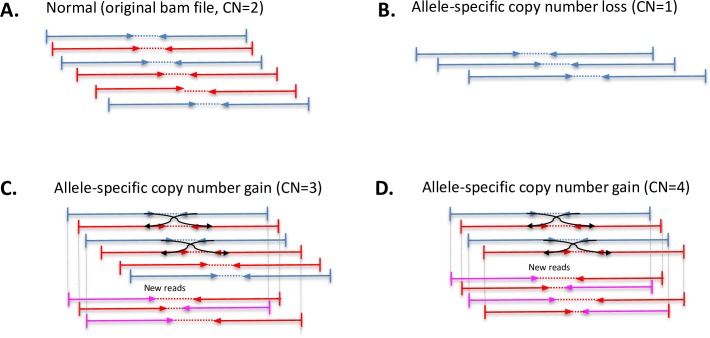
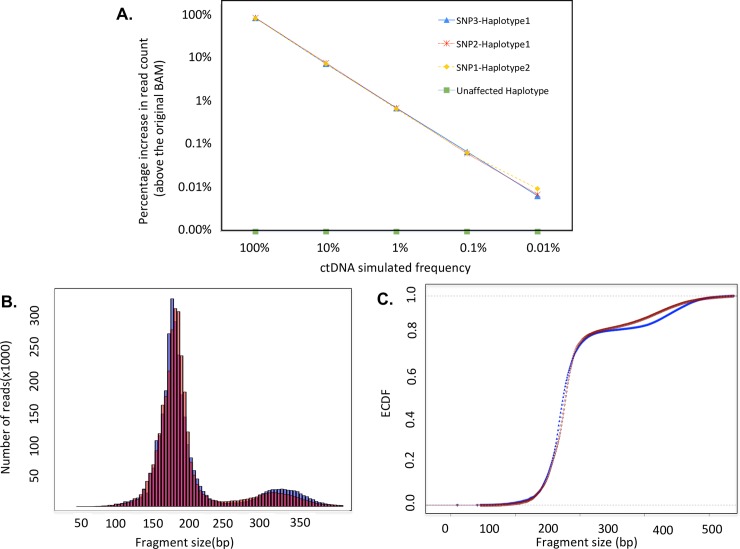
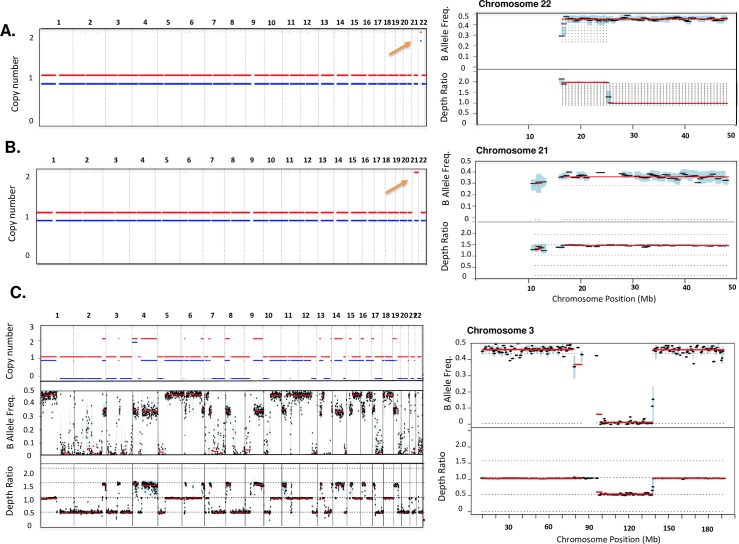
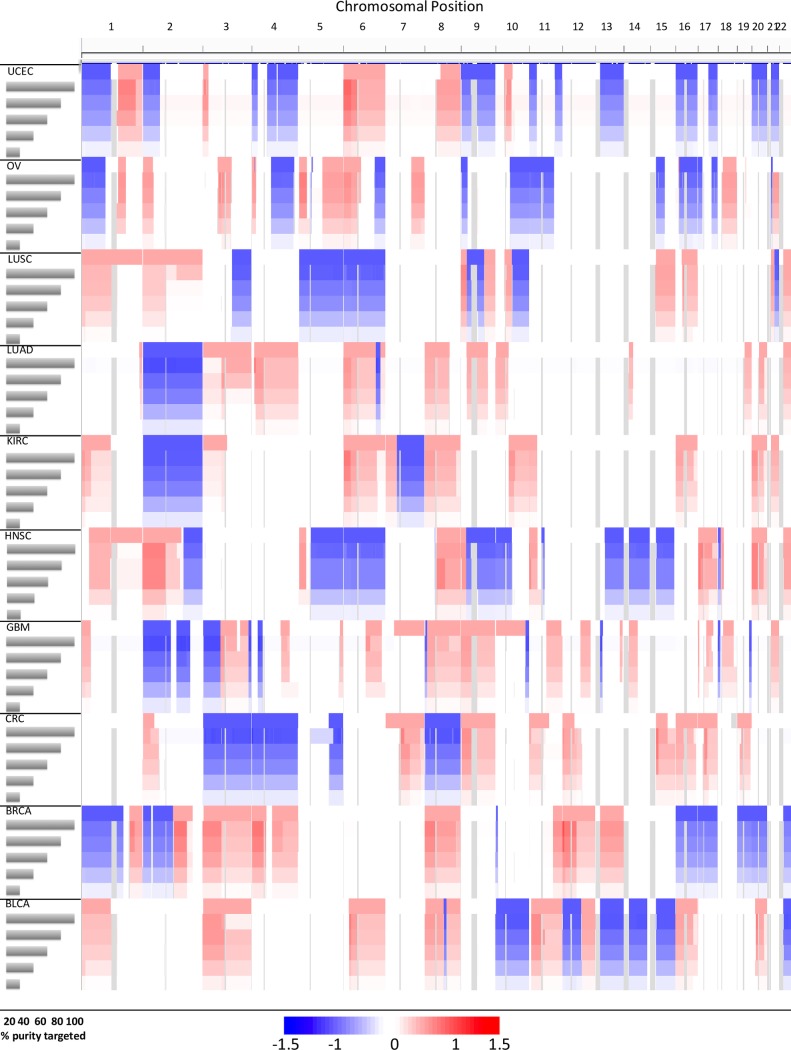
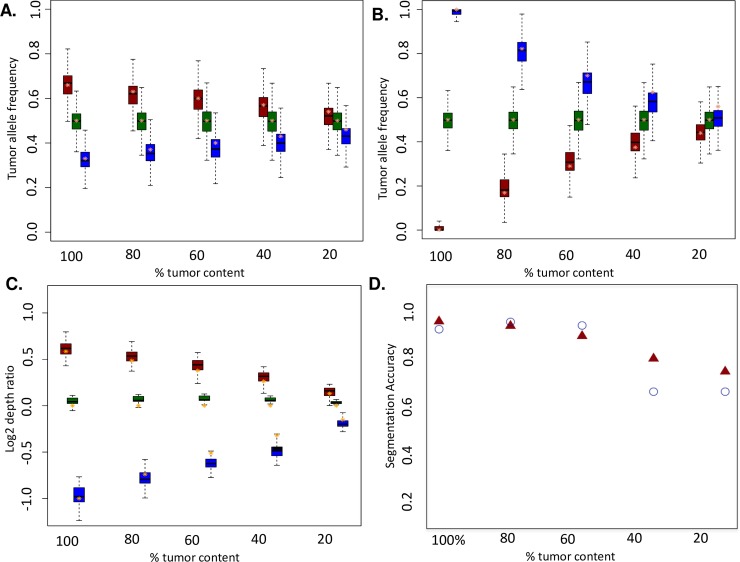
Somatic copy number variations (CNVs) play a crucial role in development of many human cancers. The broad availability of next-generation sequencing data has enabled the development of algorithms to computationally infer CNV profiles from a variety of data types including exome and targeted sequence data; currently the most prevalent types of cancer genomics data. However, systemic evaluation and comparison of these tools remains challenging due to a lack of ground truth reference sets. To address this need, we have developed Bamgineer, a tool written in Python to introduce user-defined haplotype-phased allele-specific copy number events into an existing Binary Alignment Mapping (BAM) file, with a focus on targeted and exome sequencing experiments. As input, this tool requires a read alignment file (BAM format), lists of non-overlapping genome coordinates for introduction of gains and losses (bed file), and an optional file defining known haplotypes (vcf format). To improve runtime performance, Bamgineer introduces the desired CNVs in parallel using queuing and parallel processing on a local machine or on a high-performance computing cluster. As proof-of-principle, we applied Bamgineer to a single high-coverage (mean: 220X) exome sequence file from a blood sample to simulate copy number profiles of 3 exemplar tumors from each of 10 tumor types at 5 tumor cellularity levels (20-100%, 150 BAM files in total). To demonstrate feasibility beyond exome data, we introduced read alignments to a targeted 5-gene cell-free DNA sequencing library to simulate EGFR amplifications at frequencies consistent with circulating tumor DNA (10, 1, 0.1 and 0.01%) while retaining the multimodal insert size distribution of the original data. We expect Bamgineer to be of use for development and systematic benchmarking of CNV calling algorithms by users using locally-generated data for a variety of applications. The source code is freely available at http://github.com/pughlab/bamgineer.
体细胞拷贝数变异(CNVs)在许多人类癌症的发展中起着至关重要的作用。新一代测序数据的广泛可用性使得开发算法能够从各种数据类型(包括外显子和靶向序列数据)计算推断 CNV 谱成为可能;目前这是最常见的癌症基因组学数据类型。然而,由于缺乏真实的参考数据集,对这些工具进行系统评估和比较仍然具有挑战性。为了解决这一需求,我们开发了 Bamgineer,这是一个用 Python 编写的工具,用于将用户定义的单体型相位等位基因特异性拷贝数事件引入现有的二进制对准映射(BAM)文件中,重点是靶向和外显子测序实验。作为输入,该工具需要一个读取对齐文件(BAM 格式)、用于引入增益和损耗的非重叠基因组坐标列表(bed 文件)以及定义已知单体型的可选文件(vcf 格式)。为了提高运行时性能,Bamgineer 使用队列和本地机器或高性能计算集群上的并行处理并行引入所需的 CNVs。作为原理验证,我们将 Bamgineer 应用于来自血液样本的单个高覆盖率(平均值:220X)外显子序列文件,以模拟来自 10 种肿瘤类型的每个肿瘤的 3 个肿瘤细胞活力水平(20-100%,总共 150 个 BAM 文件)的拷贝数谱。为了证明超出外显子数据的可行性,我们将读取对齐引入靶向 5 个基因的无细胞 DNA 测序文库中,以模拟与循环肿瘤 DNA 一致的 EGFR 扩增频率(10、1、0.1 和 0.01%),同时保留原始数据的多模态插入大小分布。我们预计 Bamgineer 将有助于用户使用本地生成的数据开发和系统地对 CNV 调用算法进行基准测试,以用于各种应用。源代码可在 http://github.com/pughlab/bamgineer 上免费获得。