Pimentel Juliana S M, Carmo Anderson O, Rosse Izinara C, Martins Ana P V, Ludwig Sandra, Facchin Susanne, Pereira Adriana H, Brandão-Dias Pedro F P, Abreu Nazaré L, Kalapothakis Evanguedes
Laboratory of Biotechnology and Molecular Markers, Department of General Biology, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil.
Department of Zoology, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil.
Front Genet. 2018 Mar 9;9:73. doi: 10.3389/fgene.2018.00073. eCollection 2018.
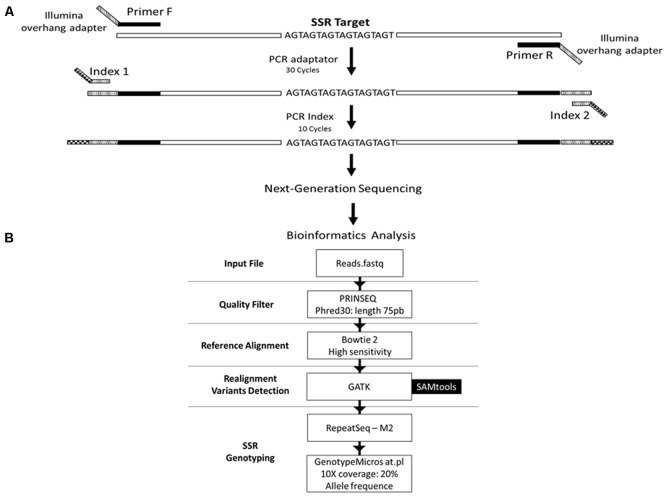
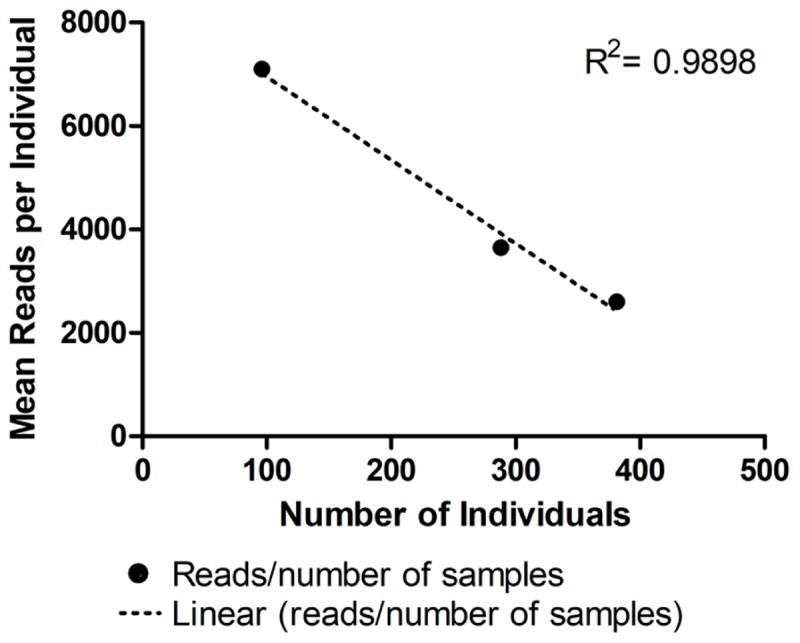
Genetic diversity and population studies are essential for conservation and wildlife management programs. However, monitoring requires the analysis of multiple from many samples. These processes can be laborious and expensive. The choice of microsatellites and PCR calibration for genotyping are particularly daunting. Here we optimized a low-cost genotyping method using multiple microsatellite for simultaneous genotyping of up to 384 samples using next-generation sequencing (NGS). We designed primers with adapters to the combinatorial barcoding amplicon library and sequenced samples by MiSeq. Next, we adapted a bioinformatics pipeline for genotyping microsatellites based on read-length and sequence content. Using primer pairs for eight microsatellite from the fish , we amplified, sequenced, and analyzed the DNA of 96, 288, or 384 individuals for allele detection. The most cost-effective methodology was a pseudo-multiplex reaction using a low-throughput kit of 1 M reads (Nano) for 384 DNA samples. We observed an average of 325 reads per individual per when genotyping eight . Assuming a minimum requirement of 10 reads per , two to four times more could be tested in each run, depending on the quality of the PCR reaction of each . In conclusion, we present a novel method for microsatellite genotyping using Illumina combinatorial barcoding that dispenses exhaustive PCR calibrations, since non-specific amplicons can be eliminated by bioinformatics analyses. This methodology rapidly provides genotyping data and is therefore a promising development for large-scale conservation-genetics studies.
遗传多样性和种群研究对于保护和野生动物管理计划至关重要。然而,监测需要对许多样本进行多个分析。这些过程可能既费力又昂贵。用于基因分型的微卫星选择和PCR校准尤其具有挑战性。在这里,我们优化了一种低成本的基因分型方法,使用多个微卫星通过下一代测序(NGS)对多达384个样本进行同时基因分型。我们设计了带有接头的引物用于组合条形码扩增子文库,并通过MiSeq对样本进行测序。接下来,我们改编了一个生物信息学流程,用于基于读长和序列内容对微卫星进行基因分型。使用来自鱼类的八个微卫星的引物对,我们扩增、测序并分析了96、288或384个个体的DNA以进行等位基因检测。最具成本效益的方法是使用1 M读长的低通量试剂盒(Nano)对384个DNA样本进行假多重反应。在对八个微卫星进行基因分型时,我们观察到每个个体平均每个微卫星有325个读长。假设每个微卫星的最低读长要求为10个,根据每个微卫星PCR反应的质量,每次运行可以测试的微卫星数量可以增加两到四倍。总之,我们提出了一种使用Illumina组合条形码进行微卫星基因分型的新方法,该方法无需进行详尽的PCR校准,因为非特异性扩增子可以通过生物信息学分析消除。这种方法能够快速提供基因分型数据,因此对于大规模保护遗传学研究来说是一个有前景的进展。