Department of Biology and Biological Engineering, Chalmers University of Technology, Gothenburg, SE, Sweden.
Wallenberg Laboratory, Department of Molecular and Clinical Medicine, University of Gothenburg, Gothenburg, SE, Sweden.
PLoS One. 2018 Mar 30;13(3):e0195161. doi: 10.1371/journal.pone.0195161. eCollection 2018.
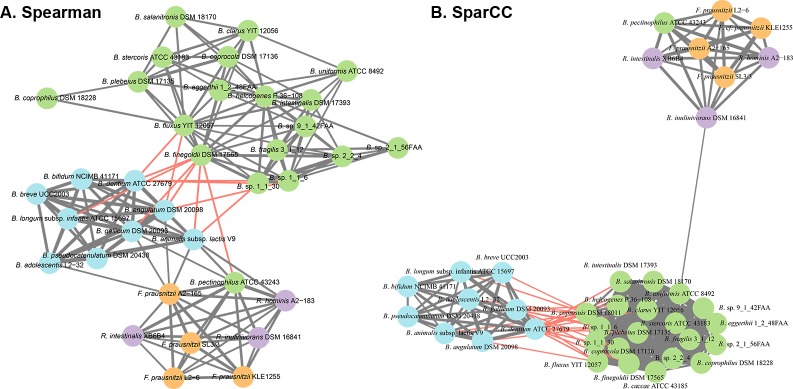
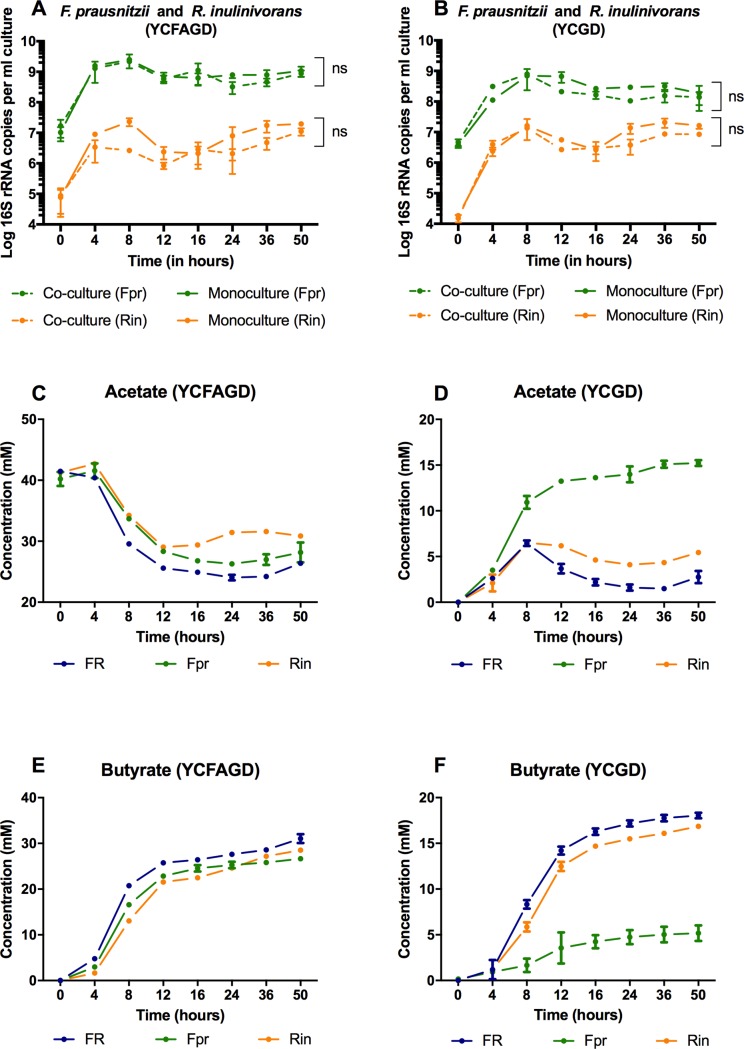
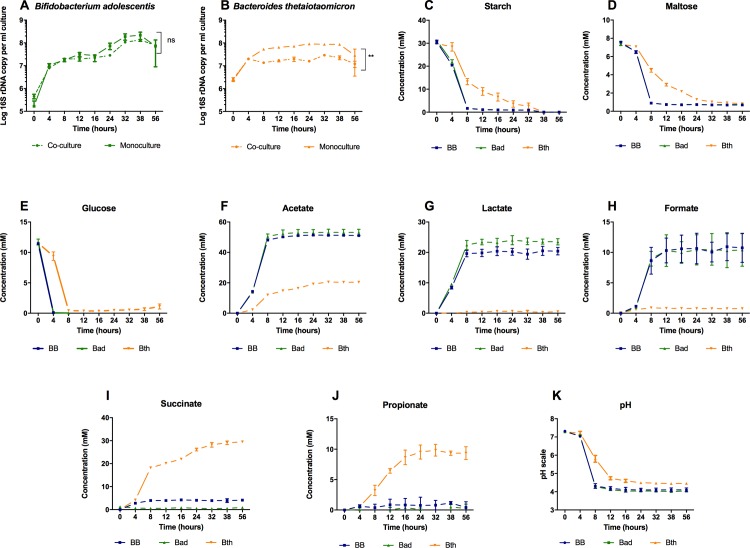
Network analysis of large metagenomic datasets generated by current sequencing technologies can reveal significant co-occurrence patterns between microbial species of a biological community. These patterns can be analyzed in terms of pairwise combinations between all species comprising a community. Here, we construct a co-occurrence network for abundant microbial species encompassing the three dominant phyla found in human gut. This was followed by an in vitro evaluation of the predicted microbe-microbe co-occurrences, where we chose species pairs Bifidobacterium adolescentis and Bacteroides thetaiotaomicron, as well as Faecalibacterium prausnitzii and Roseburia inulinivorans as model organisms for our study. We then delineate the outcome of the co-cultures when equal distributions of resources were provided. The growth behavior of the co-culture was found to be dependent on the types of microbial species present, their specific metabolic activities, and resulting changes in the culture environment. Through this reductionist approach and using novel in vitro combinations of microbial species under anaerobic conditions, the results of this work will aid in the understanding and design of synthetic community formulations.
基于当前测序技术生成的大型宏基因组数据集的网络分析可以揭示生物群落中微生物物种之间的显著共现模式。可以根据群落中包含的所有物种之间的两两组合来分析这些模式。在这里,我们构建了一个包含人类肠道中三个主要门的丰富微生物物种的共现网络。接下来,我们对预测的微生物-微生物共现进行了体外评估,选择双歧杆菌和拟杆菌以及普拉梭菌和菊粉瘤胃菌作为我们研究的模型生物。然后,我们描述了在提供均等资源分配时共培养的结果。发现共培养的生长行为取决于存在的微生物物种类型、它们特定的代谢活性以及培养环境的变化。通过这种简化方法,并在厌氧条件下使用新型微生物物种的体外组合,这项工作的结果将有助于理解和设计合成群落配方。