Laboratory of Protozoology, Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao, China.
Center for Mechanisms of Evolution, Arizona State University, Tempe, USA.
Genome Biol Evol. 2018 Mar 1;10(3):883-894. doi: 10.1093/gbe/evy055.
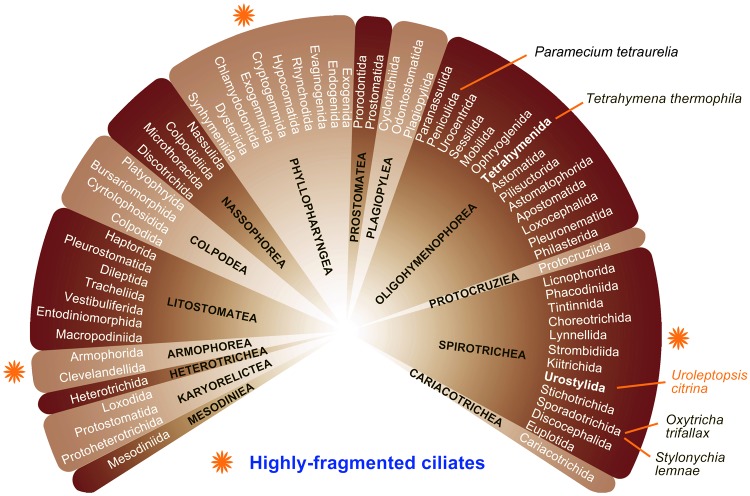
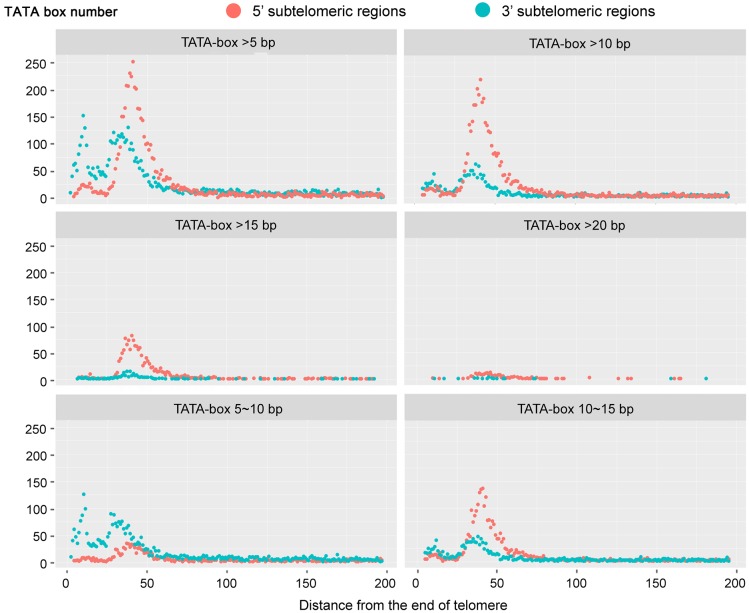
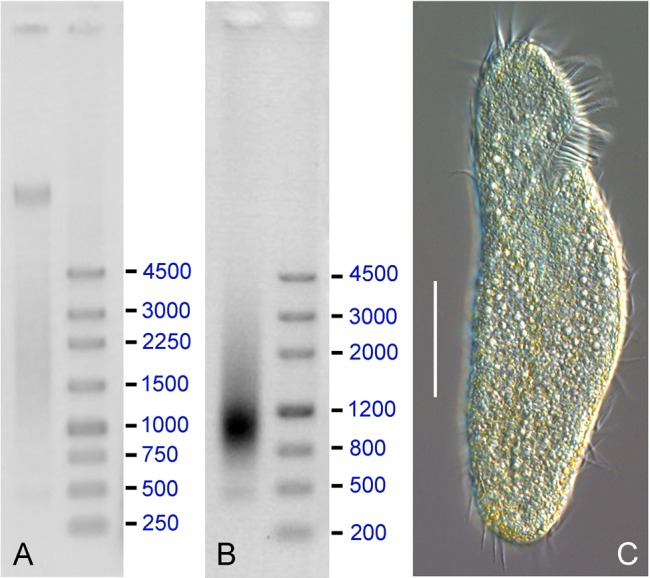
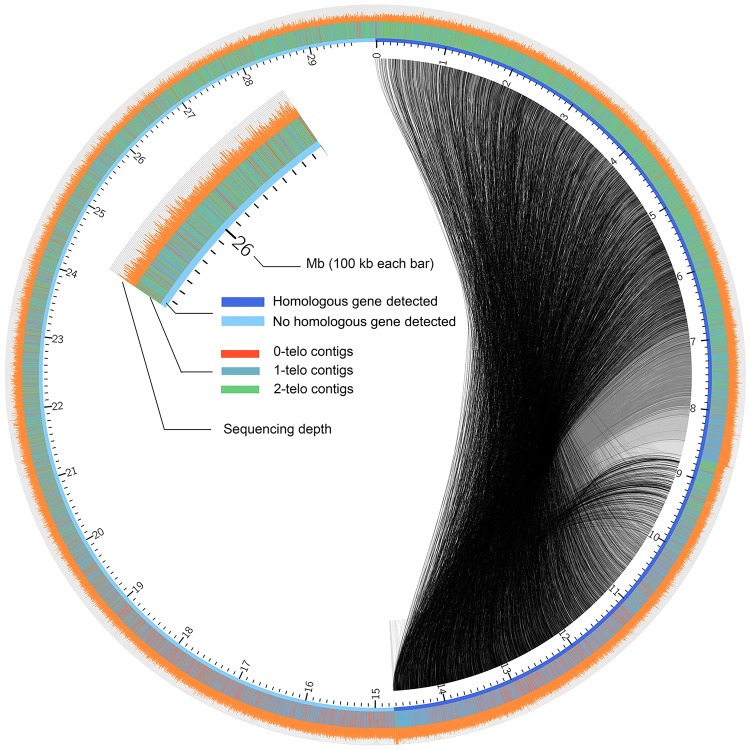
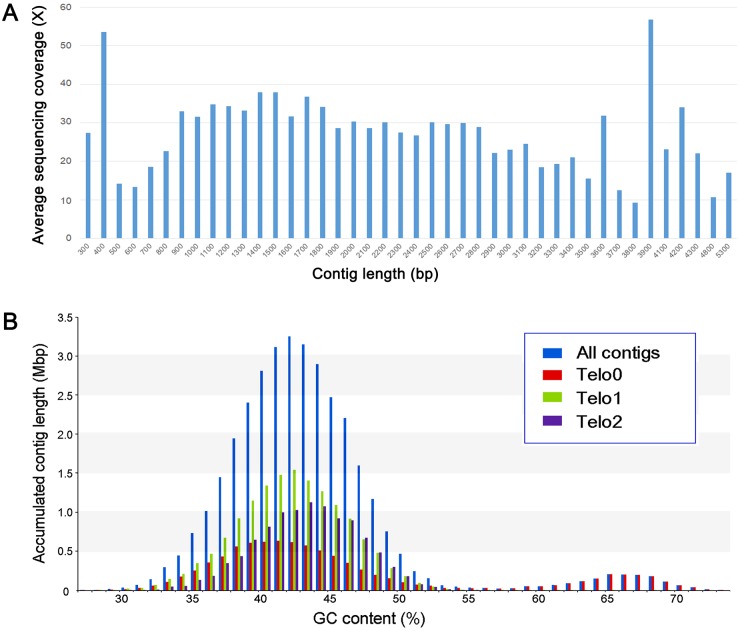
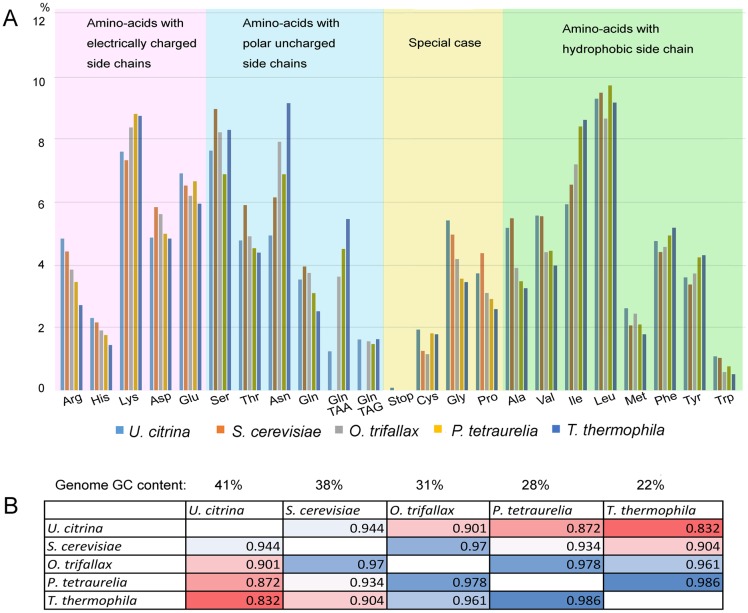
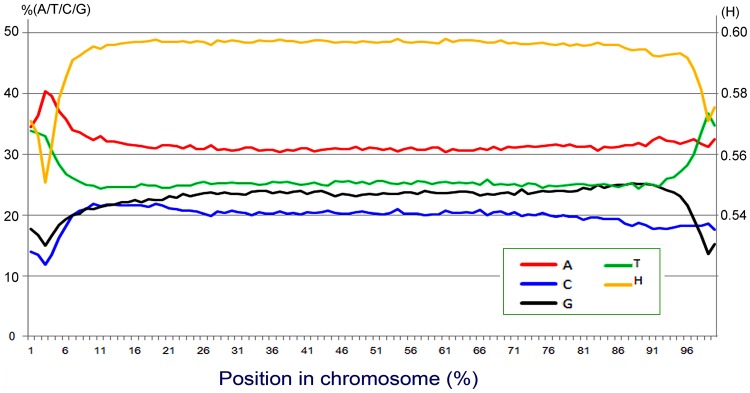
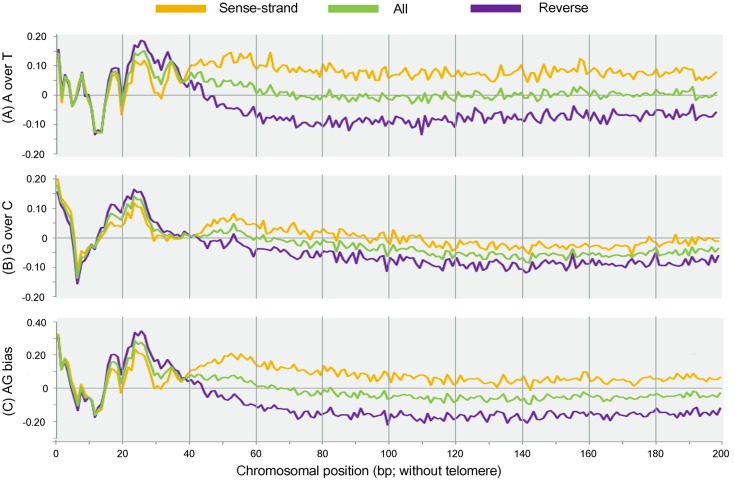
Ciliated protists are a large group of single-celled eukaryotes with separate germline and somatic nuclei in each cell. The somatic genome is developed from the zygotic nucleus through a series of chromosomal rearrangements, including fragmentation, DNA elimination, de novo telomere addition, and DNA amplification. This unique feature makes them perfect models for research in genome biology and evolution. However, genomic research of ciliates has been limited to a few species, owing to problems with DNA contamination and obstacles in cultivation. Here, we introduce a method combining telomere-primer PCR amplification and high-throughput sequencing, which can reduce DNA contamination and obtain genomic data efficiently. Based on this method, we report a draft somatic genome of a multimacronuclear ciliate, Uroleptopsis citrina. 1) The telomeric sequence in U. citrina is confirmed to be C4A4C4A4C4 by directly blunt-end cloning. 2) Genomic analysis of the resulting chromosomes shows a "one-gene one-chromosome" pattern, with a small number of multiple-gene chromosomes. 3) Amino acid usage is analyzed, and reassignment of stop codons is confirmed. 4) Chromosomal analysis shows an obvious asymmetrical GC skew and high bias between A and T in the subtelomeric regions of the sense-strand, with the detection of an 11-bp high AT motif region in the 3' subtelomeric region. 5) The subtelomeric sequence also has an obvious 40 nt strand oscillation of nucleotide ratio. 6) In the 5' subtelomeric region of the coding strand, the distribution of potential TATA-box regions is illustrated, which accumulate between 30 and 50 nt. This work provides a valuable reference for genomic research and furthers our understanding of the dynamic nature of unicellular eukaryotic genomes.
纤毛虫是一类具有单独的生殖核和体细胞核的单细胞真核生物,其体细胞基因组由合子核通过一系列染色体重排发育而来,包括碎片化、DNA 缺失、从头端粒添加和 DNA 扩增。这一独特的特征使它们成为研究基因组生物学和进化的理想模型。然而,纤毛虫的基因组研究一直受到 DNA 污染和培养障碍等问题的限制,仅限于少数几种物种。在这里,我们介绍了一种结合端粒引物 PCR 扩增和高通量测序的方法,该方法可以减少 DNA 污染并有效地获得基因组数据。基于该方法,我们报道了一种多核纤毛虫 Uroleptopsis citrina 的体细胞基因组草图。1)通过直接末端克隆,确定 U. citrina 的端粒序列为 C4A4C4A4C4。2)所得染色体的基因组分析显示出“一个基因一个染色体”的模式,少数染色体存在多个基因。3)分析了氨基酸使用情况,并确认了终止密码子的重新分配。4)染色体分析显示,在有义链的着丝粒区域,GC skew 明显不对称,A 和 T 高度偏向,在 3'着丝粒区域检测到一个 11 个碱基对的高 AT 基序区域。5)着丝粒序列也有明显的 40nt 核苷酸比链摆动。6)在编码链的 5'着丝粒区域,描绘了潜在 TATA 盒区域的分布,这些区域在 30 到 50nt 之间积累。这项工作为基因组研究提供了有价值的参考,并进一步加深了我们对单细胞真核生物基因组动态性质的理解。