Binda Anna, Rivolta Ilaria, Villa Chiara, Chisci Elisa, Beghi Massimiliano, Cornaggia Cesare M, Giovannoni Roberto, Combi Romina
School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy.
Department of Mental Health, AUSL Romagna, Ravenna, Italy.
Front Cell Neurosci. 2018 Mar 20;12:76. doi: 10.3389/fncel.2018.00076. eCollection 2018.
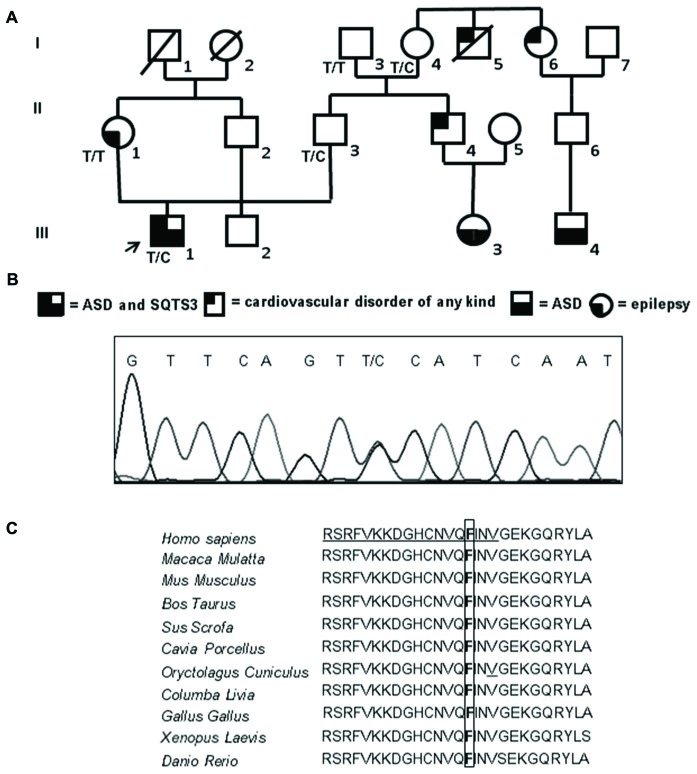
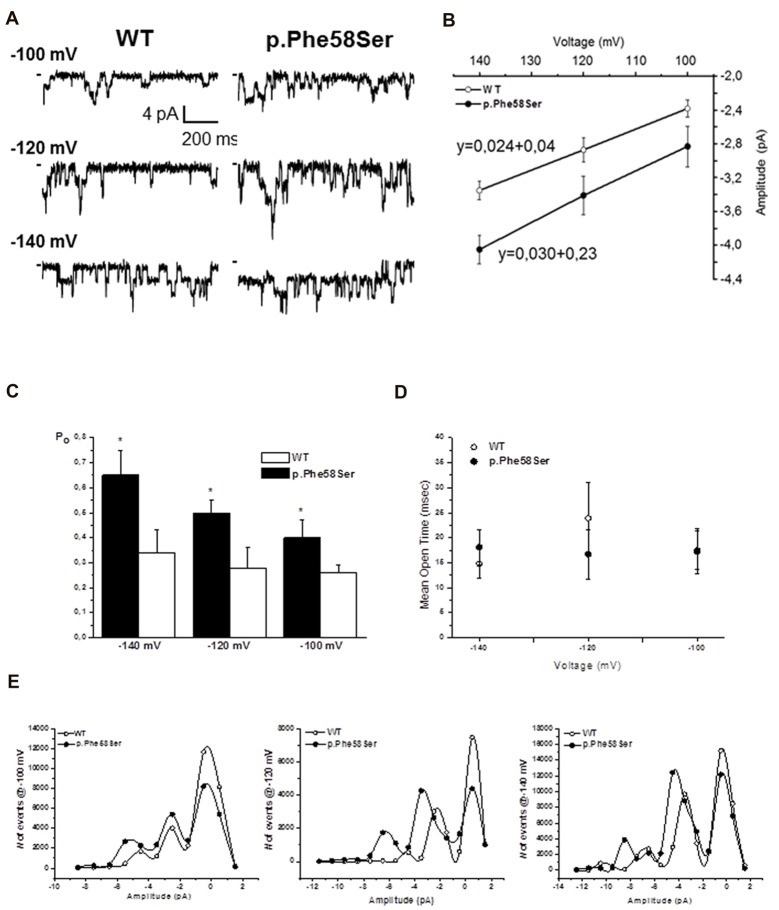
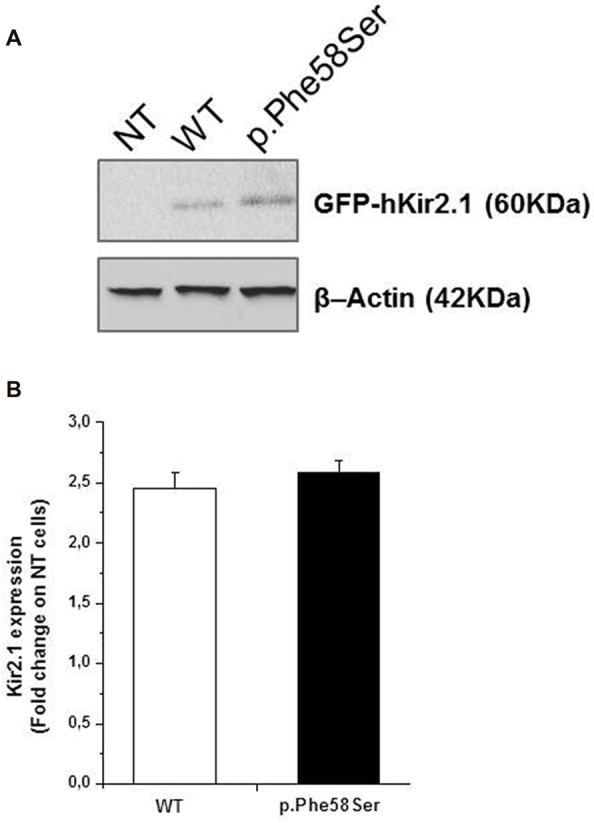
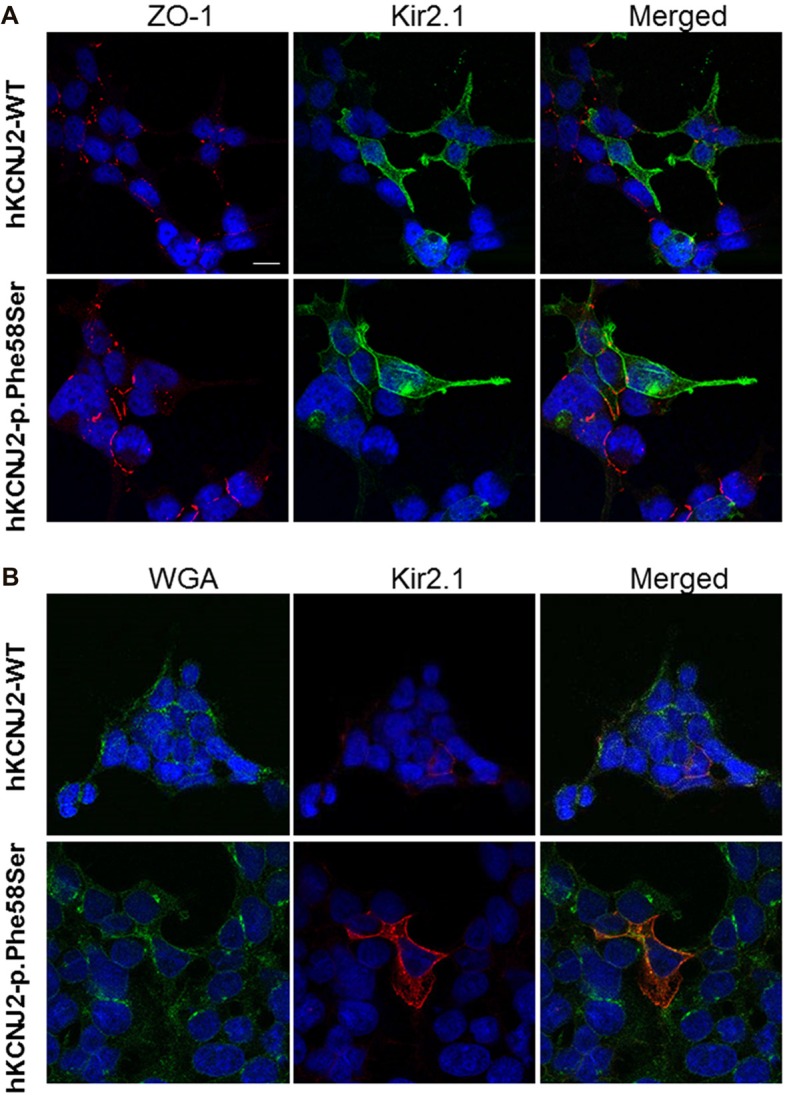
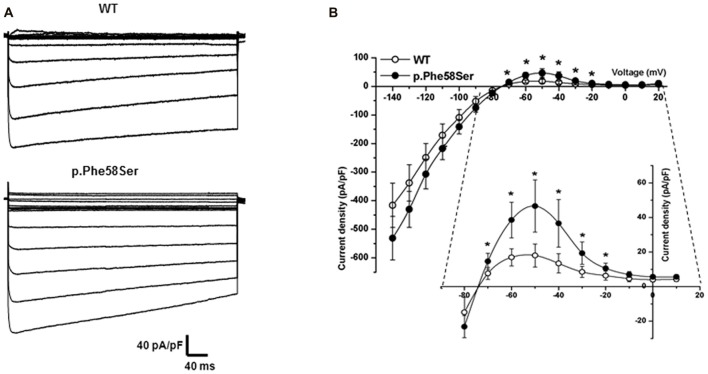
Inwardly rectifying potassium channels (Kir) have been historically associated to several cardiovascular disorders. In particular, loss-of-function mutations in the Kir2.1 channel have been reported in cases affected by Andersen-Tawil syndrome while gain-of-function mutations in the same channel cause the short QT3 syndrome. Recently, a missense mutation in Kir2.1, as well as mutations in the Kir4.1, were reported to be involved in autism spectrum disorders (ASDs) suggesting a role of potassium channels in these diseases and introducing the idea of the existence of K channel ASDs. Here, we report the identification in an Italian affected family of a novel missense mutation (p.Phe58Ser) in the gene detected in heterozygosity in a proband affected by autism and borderline for short QT syndrome type 3. The mutation is located in the N-terminal region of the gene coding for the Kir2.1 channel and in particular in a very conserved domain. assays demonstrated that this mutation results in an increase of the channel conductance and in its open probability. This gain-of-function of the protein is consistent with the autistic phenotype, which is normally associated to an altered neuronal excitability.
内向整流钾通道(Kir)在历史上一直与多种心血管疾病相关。特别是,已报道在患有安德森-陶威尔综合征的病例中存在Kir2.1通道的功能丧失突变,而同一通道的功能获得性突变则导致短QT3综合征。最近,有报道称Kir2.1中的一个错义突变以及Kir4.1中的突变与自闭症谱系障碍(ASD)有关,这表明钾通道在这些疾病中发挥作用,并引出了钾通道相关ASD存在的概念。在此,我们报告在一个意大利患病家族中鉴定出一个新的错义突变(p.Phe58Ser),该突变在一名患有自闭症且处于3型短QT综合征临界状态的先证者中以杂合子形式被检测到。该突变位于编码Kir2.1通道的基因的N端区域,特别是在一个非常保守的结构域中。实验表明,这种突变导致通道电导增加及其开放概率增加。该蛋白的这种功能获得与自闭症表型一致,自闭症表型通常与神经元兴奋性改变有关。