Queensland Alliance for Agriculture and Food Innovation, Centre for Animal Science, The University of Queensland, Building 80, 306 Carmody Road, Brisbane, St Lucia, QLD, 4072, Australia.
Collage of Science, Health and Engineering, La Trobe University, Melbourne, VIC, 3086, Australia.
BMC Genomics. 2018 Apr 5;19(1):237. doi: 10.1186/s12864-018-4617-x.
There are an exceedingly large number of sequence variants discovered through whole genome sequencing in most populations, including cattle. Deciphering which of these affect complex traits is a major challenge. In this study we hypothesize that variants in some functional classes, such as splice site regions, coding regions, DNA methylated regions and long noncoding RNA will explain more variance in complex traits than others. Two variance component approaches were used to test this hypothesis - the first determines if variants in a functional class capture a greater proportion of the variance, than expected by chance, the second uses the proportion of variance explained when variants in all annotations are fitted simultaneously.
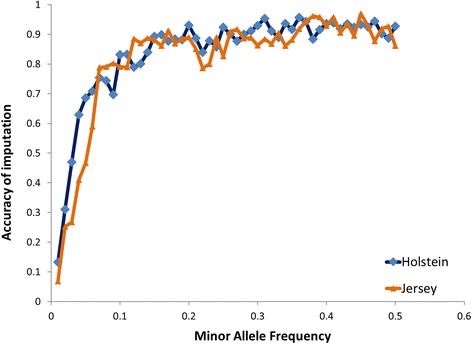
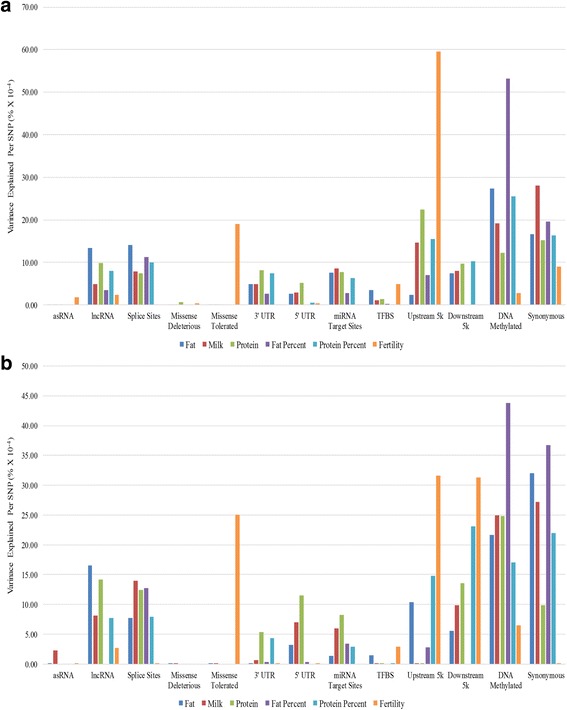
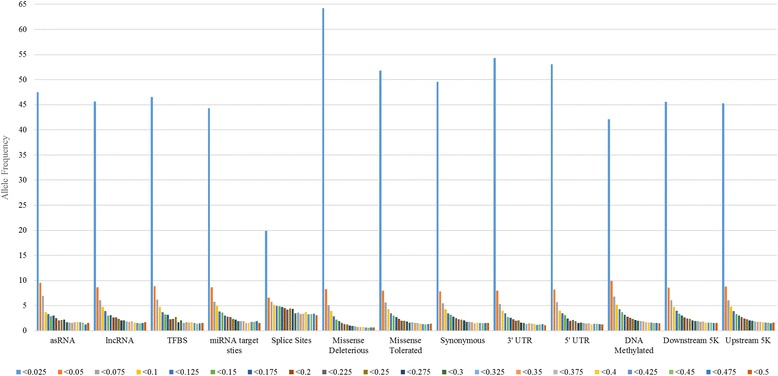
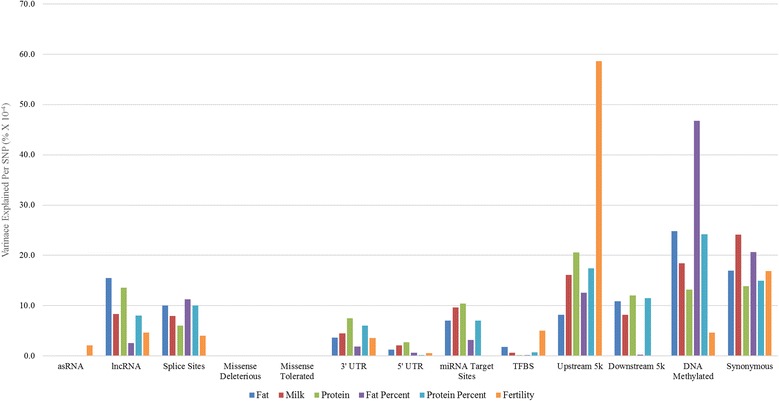
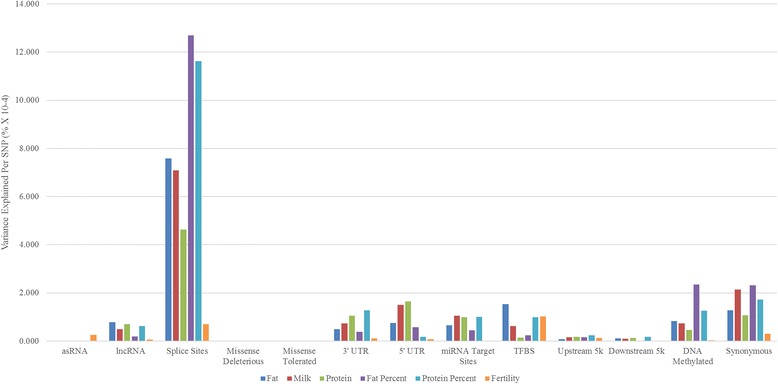
Our data set consisted of 28.3 million imputed whole genome sequence variants in 16,581 dairy cattle with records for 6 complex trait phenotypes, including production and fertility. We found that sequence variants in splice site regions and synonymous classes captured the greatest proportion of the variance, explaining up to 50% of the variance across all traits. We also found sequence variants in target sites for DNA methylation (genomic regions that are found be highly methylated in bovine placentas), captured a significant proportion of the variance. Per sequence variant, splice site variants explain the highest proportion of variance in this study. The proportion of variance captured by the missense predicted deleterious (from SIFT) and missense tolerated classes was relatively small.
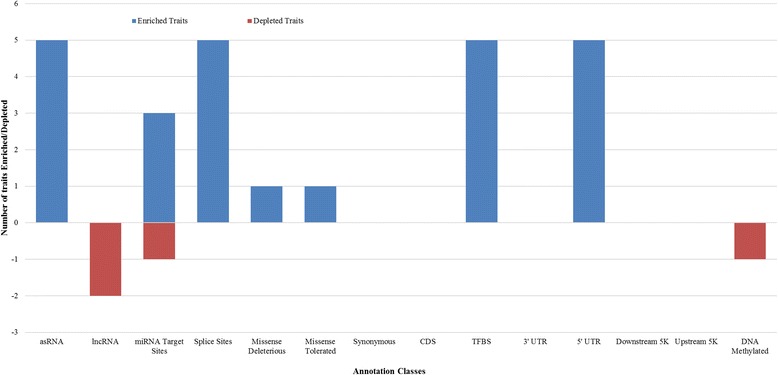
The results demonstrate using functional annotations to filter whole genome sequence variants into more informative subsets could be useful for prioritization of the variants that are more likely to be associated with complex traits. In addition to variants found in splice sites and protein coding genes regulatory variants and those found in DNA methylated regions, explained considerable variation in milk production and fertility traits. In our analysis synonymous variants captured a significant proportion of the variance, which raises the possible explanation that synonymous mutations might have some effects, or more likely that these variants are miss-annotated, or alternatively the results reflect imperfect imputation of the actual causative variants.
在大多数人群中,包括牛在内,通过全基因组测序发现了大量的序列变异。 破译哪些变异会影响复杂性状是一个主要的挑战。 在这项研究中,我们假设某些功能类别的变异,如剪接位点区域、编码区域、DNA 甲基化区域和长非编码 RNA 的变异,比其他变异能解释更多的复杂性状变异。 我们使用两种方差成分方法来检验这一假设 - 第一种方法是确定功能类别中的变异是否比随机预期捕获更多的变异,第二种方法是使用所有注释中的变异同时拟合时解释的变异比例。
我们的数据集中包含了 16581 头奶牛的 2830 万个全基因组序列变异,这些奶牛有 6 种复杂表型的记录,包括生产和繁殖。 我们发现,剪接位点区域和同义类别的序列变异捕获了最大比例的变异,解释了所有性状中高达 50%的变异。 我们还发现,DNA 甲基化的靶位点(在牛胎盘高度甲基化的基因组区域)的序列变异也捕获了相当大的一部分变异。 每个序列变异,剪接位点变异在本研究中解释了最高比例的变异。 错义预测有害(来自 SIFT)和错义耐受类别的变异所捕获的方差比例相对较小。
结果表明,使用功能注释将全基因组序列变异过滤为更具信息量的子集,可能有助于优先考虑与复杂性状更相关的变异。 除了在剪接位点和蛋白质编码基因调控区以及在 DNA 甲基化区域发现的变异外,还解释了产奶量和繁殖性状的大量变异。 在我们的分析中,同义变异捕获了相当大的一部分变异,这可能表明同义突变可能有一些影响,或者更可能是这些变异被错误注释,或者结果反映了实际致病变异的不完全推断。