Department of Biomedical Sciences, Texas A&M University College of Dentistry, Dallas, TX 75246, United States.
Department of Biomedical Sciences, Texas A&M University College of Dentistry, Dallas, TX 75246, United States.
Bone. 2018 Jul;112:71-89. doi: 10.1016/j.bone.2018.03.027. Epub 2018 Apr 5.
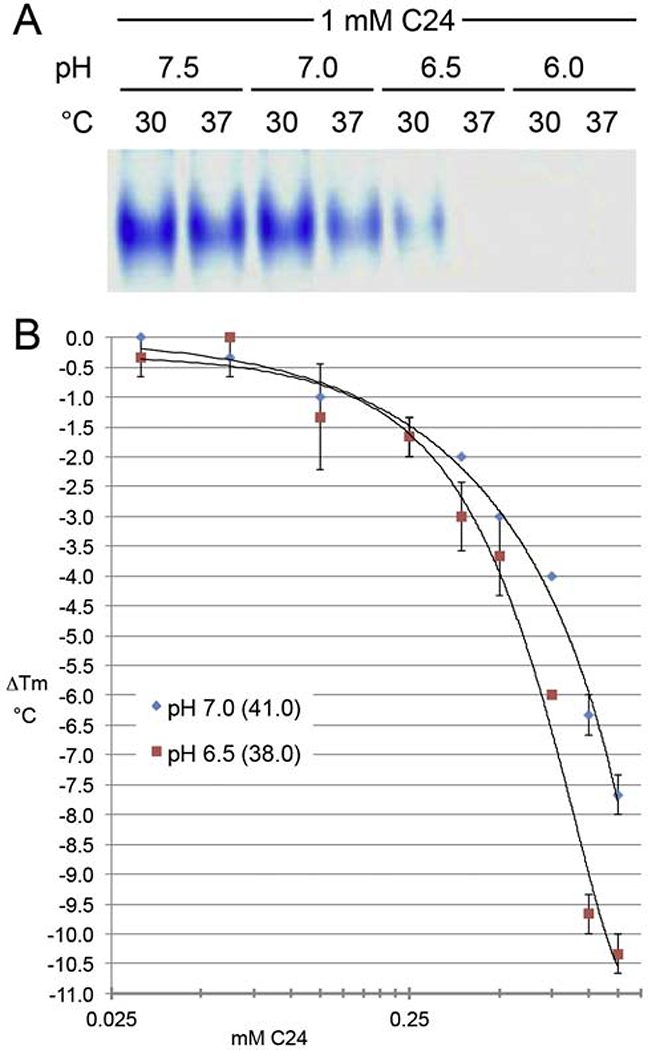
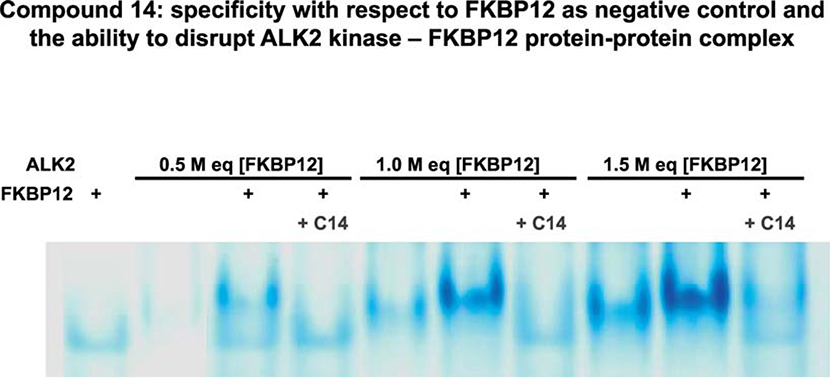
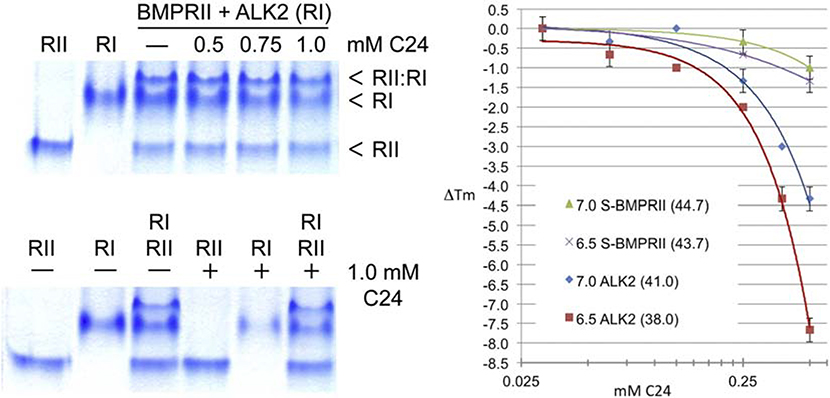
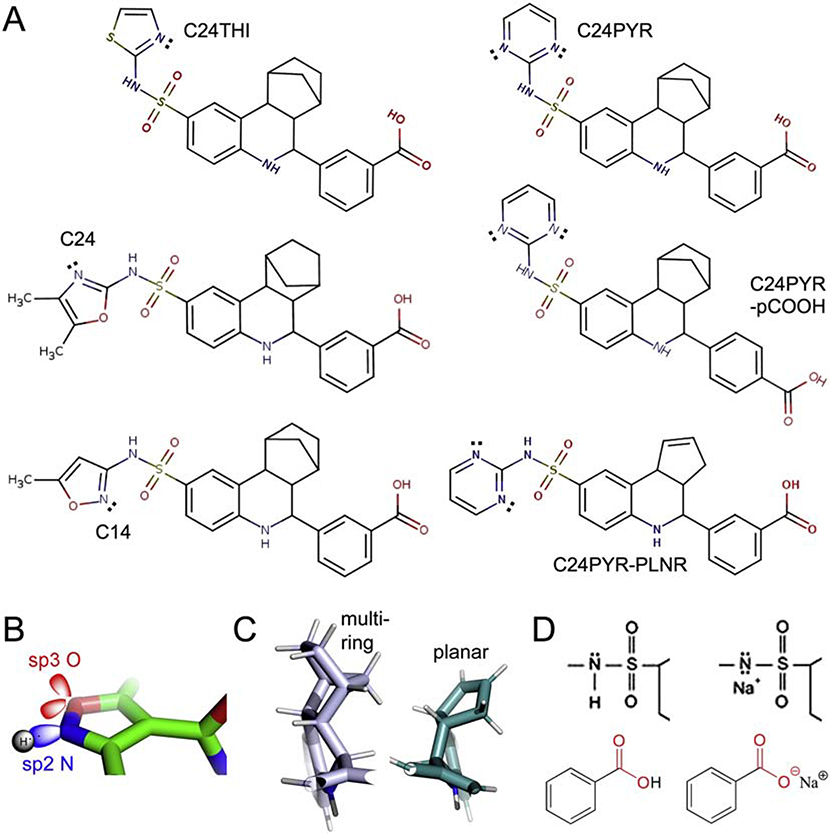
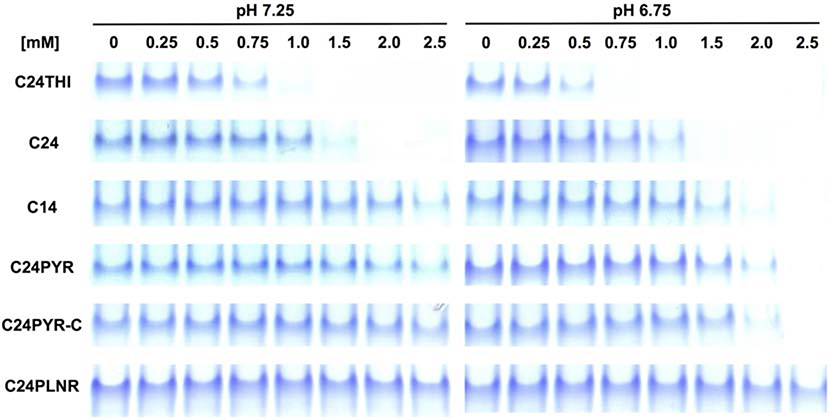
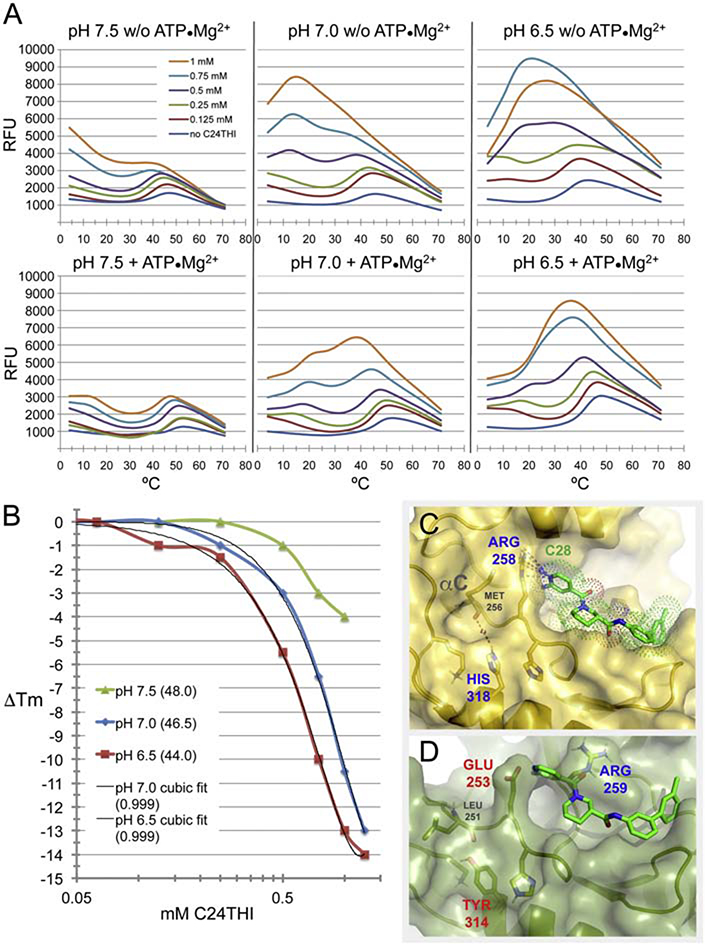
Heterotopic ossification (HO), the pathological extraskeletal formation of bone, can arise from blast injuries, severe burns, orthopedic procedures and gain-of-function mutations in a component of the bone morphogenetic protein (BMP) signaling pathway, the ACVR1/ALK2 receptor serine-threonine (protein) kinase, causative of Fibrodysplasia Ossificans Progressiva (FOP). All three ALKs (-2, -3, -6) that play roles in bone morphogenesis contribute to trauma-induced HO, hence are well-validated pharmacological targets. That said, development of inhibitors, typically competitors of ATP binding, is inherently difficult due to the conserved nature of the active site of the 500+ human protein kinases. Since these enzymes are regulated via inherent plasticity, pharmacological chaperone-like drugs binding to another (allosteric) site could hypothetically modulate kinase conformation and activity. To test for such a mechanism, a surface pocket of ALK2 kinase formed largely by a key allosteric substructure was targeted by supercomputer docking of drug-like compounds from a virtual library. Subsequently, the effects of docked hits were further screened in vitro with purified recombinant kinase protein. A family of compounds with terminal hydrogen-bonding acceptor groups was identified that significantly destabilized the protein, inhibiting activity. Destabilization was pH-dependent, putatively mediated by ionization of a histidine within the allosteric substructure with decreasing pH. In vivo, nonnative proteins are degraded by proteolysis in the proteasome complex, or cellular trashcan, allowing for the emergence of therapeutics that inhibit through degradation of over-active proteins implicated in the pathology of diseases and disorders. Because HO is triggered by soft-tissue trauma and ensuing hypoxia, dependency of ALK destabilization on hypoxic pH imparts selective efficacy on the allosteric inhibitors, providing potential for safe prophylactic use.
异位骨化(HO)是一种病理性的骨骼外骨形成,可以由爆炸伤、严重烧伤、骨科手术和骨形态发生蛋白(BMP)信号通路的组成部分——ACVR1/ALK2 受体丝氨酸-苏氨酸(蛋白)激酶的功能获得性突变引起,导致纤维性骨发育不良(FOP)。在骨骼形态发生中发挥作用的所有三种 ALK(-2、-3、-6)都有助于创伤诱导的 HO,因此是经过充分验证的药理学靶点。也就是说,由于 500 多种人类蛋白激酶的活性位点具有保守性,因此开发通常与 ATP 结合竞争的抑制剂具有内在的困难。由于这些酶通过固有可塑性进行调节,因此与另一个(变构)位点结合的药理学伴侣样药物可能可以假设调节激酶构象和活性。为了测试这种机制,ALK2 激酶的一个表面口袋主要由一个关键的变构亚结构形成,通过来自虚拟文库的药物样化合物的超级计算机对接来靶向该口袋。随后,通过用纯化的重组激酶蛋白进行体外筛选来进一步筛选对接命中物。鉴定出一组具有末端氢键受体基团的化合物,这些化合物显著破坏了蛋白质,抑制了其活性。失稳依赖于 pH 值,推测是通过变构亚结构中的组氨酸在 pH 值降低时的离子化介导的。在体内,未成熟的蛋白质通过蛋白酶体复合物或细胞垃圾桶中的蛋白水解降解,从而产生通过降解在疾病和紊乱的病理中起作用的过表达蛋白来抑制的治疗药物。由于 HO 是由软组织创伤和随之而来的缺氧触发的,ALK 的失稳依赖于缺氧 pH 值,这赋予了变构抑制剂的选择性疗效,为安全的预防性使用提供了潜力。