Chang Pearl, Gohain Moloya, Yen Ming-Ren, Chen Pao-Yang
Institute of Plant and Microbial Biology, Academia Sinica, Taipei, Taiwan.
Comput Struct Biotechnol J. 2018 Feb 15;16:43-53. doi: 10.1016/j.csbj.2018.02.003. eCollection 2018.
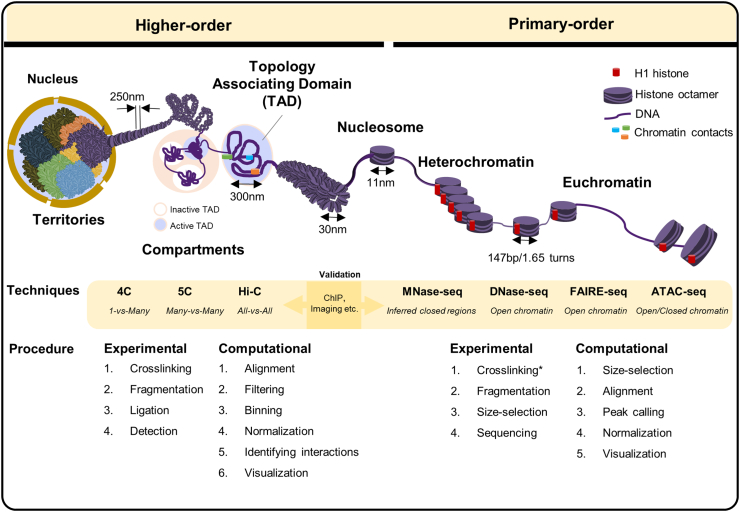
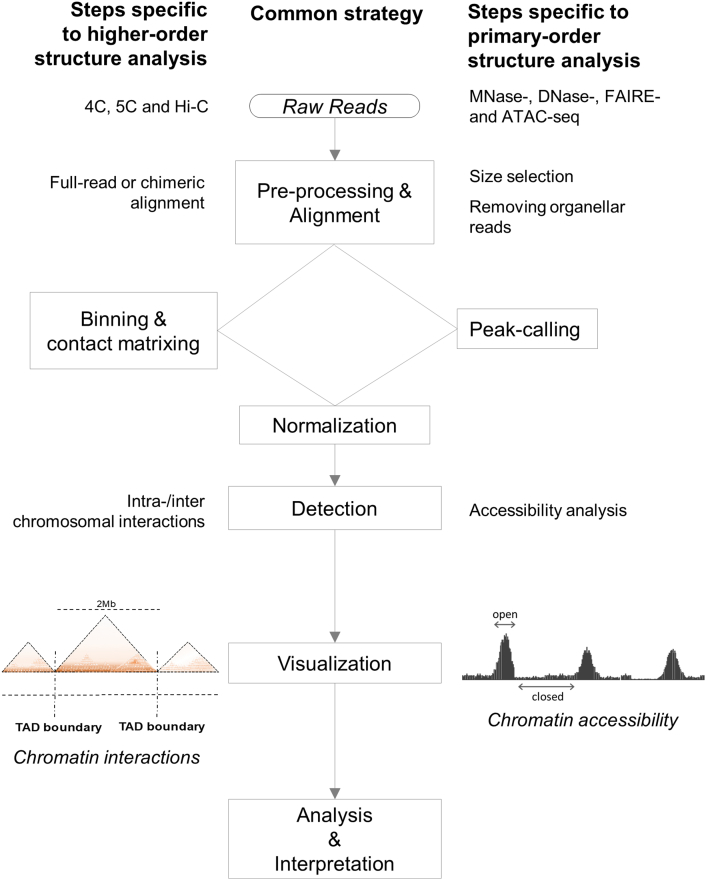
The hierarchical organization of chromatin is known to associate with diverse cellular functions; however, the precise mechanisms and the 3D structure remain to be determined. With recent advances in high-throughput next generation sequencing (NGS) techniques, genome-wide profiling of chromatin structures is made possible. Here, we provide a comprehensive overview of NGS-based methods for profiling "higher-order" and "primary-order" chromatin structures from both experimental and computational aspects. Experimental requirements and considerations specific for each method were highlighted. For computational analysis, we summarized a common analysis strategy for both levels of chromatin assessment, focusing on the characteristic computing steps and the tools. The recently developed single-cell level techniques based on Hi-C and ATAC-seq present great potential to reveal cell-to-cell variability in chromosome architecture. A brief discussion on these methods in terms of experimental and data analysis features is included. We also touch upon the biological relevance of chromatin organization and how the combination with other techniques uncovers the underlying mechanisms. We conclude with a summary and our prospects on necessary improvements of currently available methods in order to advance understanding of chromatin hierarchy. Our review brings together the analyses of both higher- and primary-order chromatin structures, and serves as a roadmap when choosing appropriate experimental and computational methods for assessing chromatin hierarchy.
染色质的分层组织与多种细胞功能相关;然而,其精确机制和三维结构仍有待确定。随着高通量下一代测序(NGS)技术的最新进展,全基因组染色质结构分析成为可能。在此,我们从实验和计算两个方面全面概述基于NGS的“高阶”和“初级”染色质结构分析方法。突出了每种方法的实验要求和注意事项。对于计算分析,我们总结了染色质评估两个层面的通用分析策略,重点关注特征计算步骤和工具。最近基于Hi-C和ATAC-seq开发的单细胞水平技术在揭示染色体结构的细胞间变异性方面具有巨大潜力。本文简要讨论了这些方法在实验和数据分析方面的特点。我们还探讨了染色质组织的生物学相关性以及与其他技术结合如何揭示潜在机制。我们最后总结并展望了当前可用方法为推进对染色质层次结构的理解而进行必要改进的方向。我们的综述汇集了对高阶和初级染色质结构的分析,并为选择评估染色质层次结构的合适实验和计算方法提供了路线图。