Zhong Wujuan, Liu Weifang, Chen Jiawen, Sun Quan, Hu Ming, Li Yun
Biostatistics and Research Decision Sciences, Merck & Co, Inc, Rahway, NJ, United States.
Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States.
Front Cell Dev Biol. 2022 Aug 19;10:957292. doi: 10.3389/fcell.2022.957292. eCollection 2022.
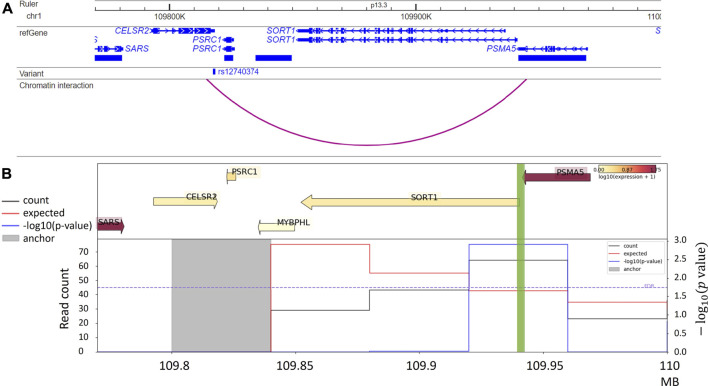
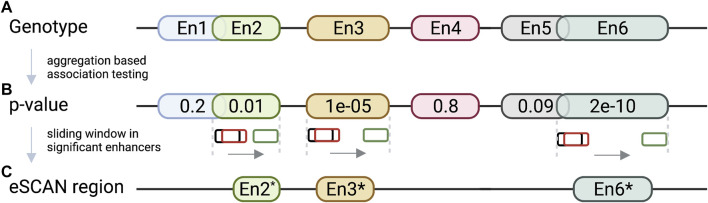
Genome-wide association studies (GWAS) have identified a vast number of variants associated with various complex human diseases and traits. However, most of these GWAS variants reside in non-coding regions producing no proteins, making the interpretation of these variants a daunting challenge. Prior evidence indicates that a subset of non-coding variants detected within or near cis-regulatory elements (e.g., promoters, enhancers, silencers, and insulators) might play a key role in disease etiology by regulating gene expression. Advanced sequencing- and imaging-based technologies, together with powerful computational methods, enabling comprehensive characterization of regulatory DNA interactions, have substantially improved our understanding of the three-dimensional (3D) genome architecture. Recent literature witnesses plenty of examples where using chromosome conformation capture (3C)-based technologies successfully links non-coding variants to their target genes and prioritizes relevant tissues or cell types. These examples illustrate the critical capability of 3D genome organization in annotating non-coding GWAS variants. This review discusses how 3D genome organization information contributes to elucidating the potential roles of non-coding GWAS variants in disease etiology.
全基因组关联研究(GWAS)已经鉴定出大量与各种复杂人类疾病和性状相关的变异。然而,这些GWAS变异大多位于不产生蛋白质的非编码区域,这使得对这些变异的解读成为一项艰巨的挑战。先前的证据表明,在顺式调控元件(如启动子、增强子、沉默子和绝缘子)内部或附近检测到的一部分非编码变异可能通过调节基因表达在疾病病因学中发挥关键作用。先进的基于测序和成像的技术,连同强大的计算方法,能够全面表征调控DNA相互作用,极大地增进了我们对三维(3D)基因组结构的理解。最近的文献中有大量例子表明,使用基于染色体构象捕获(3C)的技术成功地将非编码变异与其靶基因联系起来,并对相关组织或细胞类型进行了优先级排序。这些例子说明了3D基因组组织在注释非编码GWAS变异方面的关键能力。本综述讨论了3D基因组组织信息如何有助于阐明非编码GWAS变异在疾病病因学中的潜在作用。