IMPACT Strategic Research Centre, School of Medicine, Barwon Health, Deakin University, P.O. Box 291, Geelong, Victoria, Australia.
Department of Psychiatry, Level 1 North, Main Block, Royal Melbourne Hospital, University of Melbourne, Parkville, Victoria, Australia.
Mol Neurobiol. 2019 Jan;56(1):406-434. doi: 10.1007/s12035-018-1092-y. Epub 2018 Apr 29.
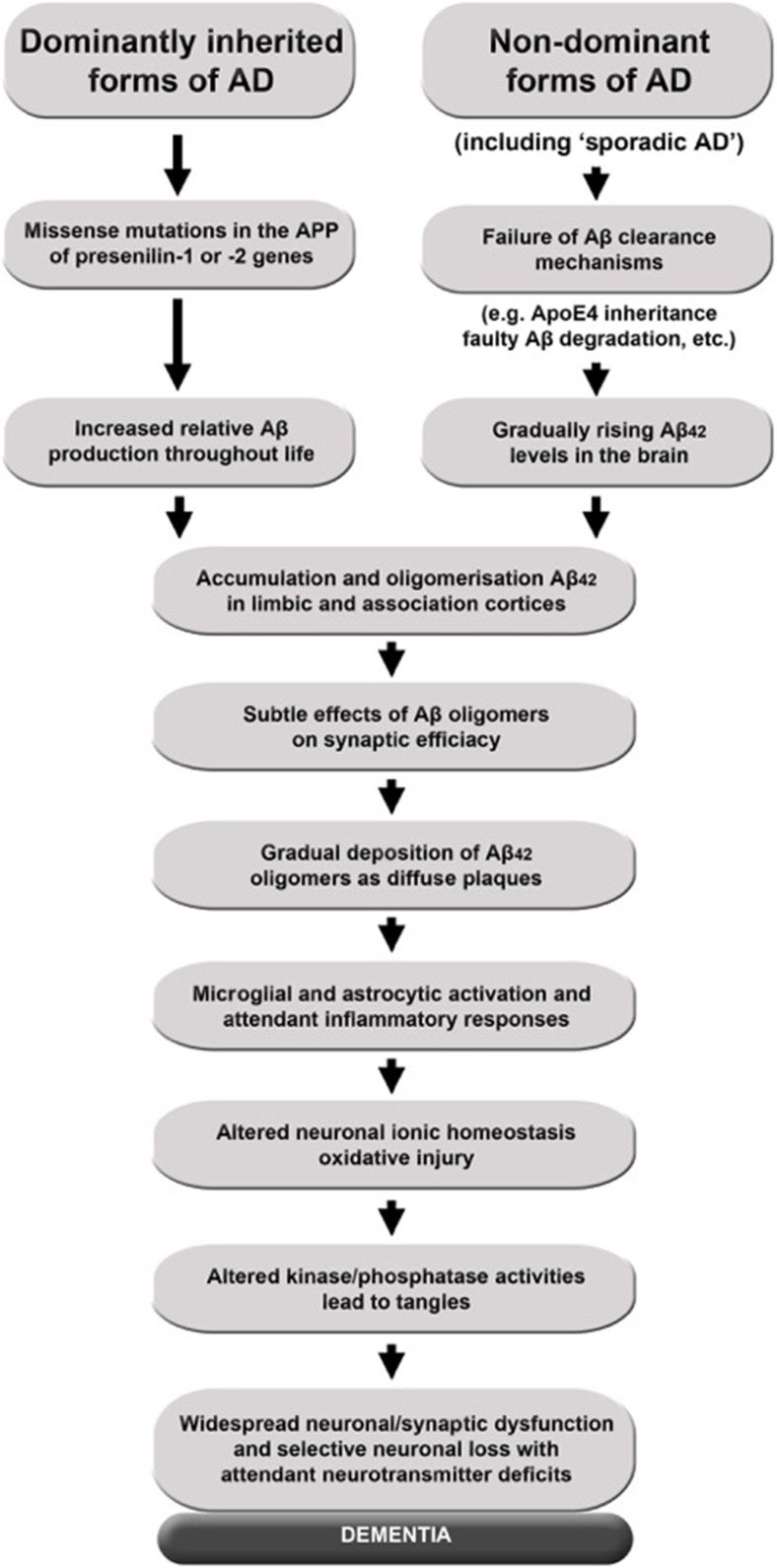
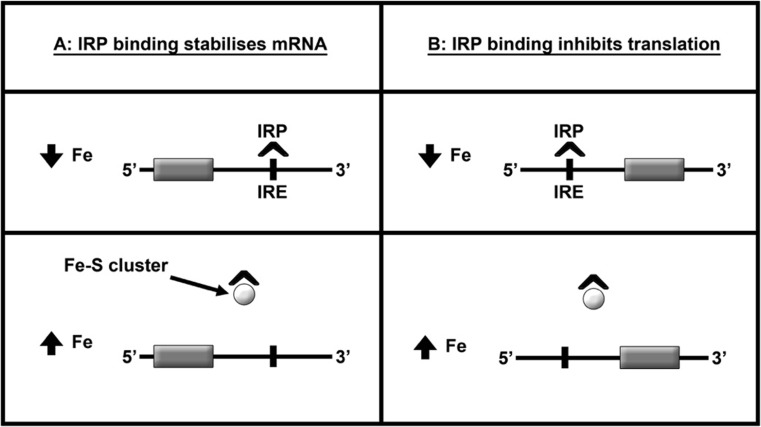
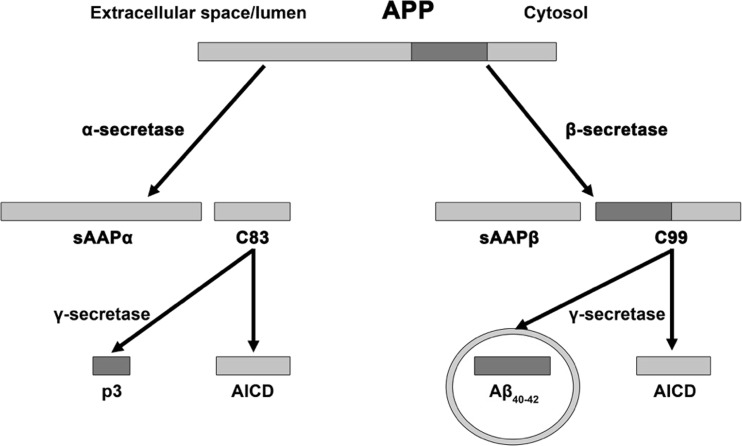
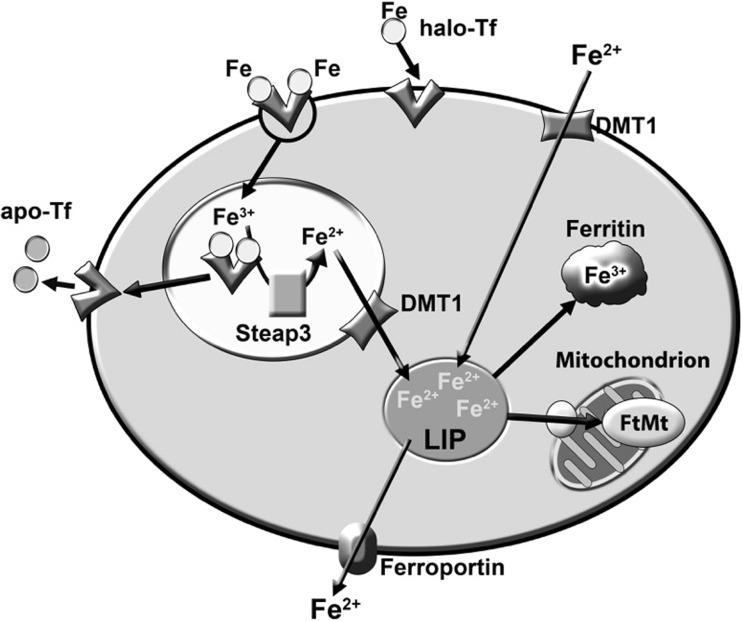
The classical amyloid cascade model for Alzheimer's disease (AD) has been challenged by several findings. Here, an alternative molecular neurobiological model is proposed. It is shown that the presence of the APOE ε4 allele, altered miRNA expression and epigenetic dysregulation in the promoter region and exon 1 of TREM2, as well as ANK1 hypermethylation and altered levels of histone post-translational methylation leading to increased transcription of TNFA, could variously explain increased levels of peripheral and central inflammation found in AD. In particular, as a result of increased activity of triggering receptor expressed on myeloid cells 2 (TREM-2), the presence of the apolipoprotein E4 (ApoE4) isoform, and changes in ANK1 expression, with subsequent changes in miR-486 leading to altered levels of protein kinase B (Akt), mechanistic (previously mammalian) target of rapamycin (mTOR) and signal transducer and activator of transcription 3 (STAT3), all of which play major roles in microglial activation, proliferation and survival, there is activation of microglia, leading to the subsequent (further) production of cytokines, chemokines, nitric oxide, prostaglandins, reactive oxygen species, inducible nitric oxide synthase and cyclooxygenase-2, and other mediators of inflammation and neurotoxicity. These changes are associated with the development of amyloid and tau pathology, mitochondrial dysfunction (including impaired activity of the electron transport chain, depleted basal mitochondrial potential and oxidative damage to key tricarboxylic acid enzymes), synaptic dysfunction, altered glycogen synthase kinase-3 (GSK-3) activity, mTOR activation, impairment of autophagy, compromised ubiquitin-proteasome system, iron dyshomeostasis, changes in APP translation, amyloid plaque formation, tau hyperphosphorylation and neurofibrillary tangle formation.
阿尔茨海默病(AD)的经典淀粉样蛋白级联模型受到了一些发现的挑战。在这里,提出了一种替代的分子神经生物学模型。研究表明,APOE ε4 等位基因的存在、TREM2 启动子区域和外显子 1 中 miRNA 表达的改变和表观遗传失调、ANK1 过度甲基化以及组蛋白翻译后甲基化水平的改变,导致 TNF-α 的转录增加,都可以解释 AD 中发现的外周和中枢炎症水平的增加。特别是,由于髓样细胞表达的触发受体 2(TREM-2)的活性增加、载脂蛋白 E4(ApoE4)同工型的存在以及 ANK1 表达的改变,随后导致 miR-486 水平的改变,从而改变蛋白激酶 B(Akt)、机械(先前的哺乳动物)雷帕霉素靶蛋白(mTOR)和信号转导和转录激活因子 3(STAT3)的水平,所有这些都在小胶质细胞的激活、增殖和存活中发挥主要作用,导致小胶质细胞的激活,进而(进一步)产生细胞因子、趋化因子、一氧化氮、前列腺素、活性氧、诱导型一氧化氮合酶和环氧化酶-2 以及炎症和神经毒性的其他介质。这些变化与淀粉样蛋白和 tau 病理学的发展、线粒体功能障碍(包括电子传递链活性受损、基础线粒体电位耗竭以及关键三羧酸酶的氧化损伤)、突触功能障碍、糖原合酶激酶-3(GSK-3)活性改变、mTOR 激活、自噬受损、泛素-蛋白酶体系统受损、铁动态平衡失调、APP 翻译改变、淀粉样斑块形成、tau 过度磷酸化和神经原纤维缠结形成有关。