Al-Ghalith Gabriel A, Hillmann Benjamin, Ang Kaiwei, Shields-Cutler Robin, Knights Dan
Bioinformatics and Computational Biology, University of Minnesota-Twin Cities, Minneapolis, Minnesota, USA.
Computer Science, University of Minnesota-Twin Cities, Minneapolis, Minnesota, USA.
mSystems. 2018 Apr 24;3(3). doi: 10.1128/mSystems.00202-17. eCollection 2018 May-Jun.
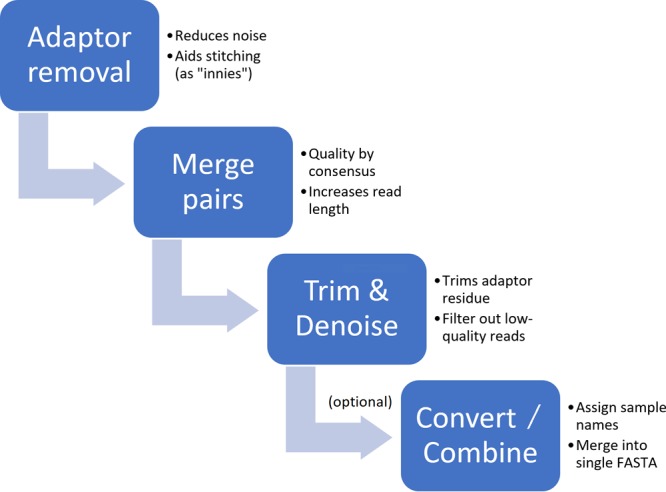
Next-generation sequencing technology is of great importance for many biological disciplines; however, due to technical and biological limitations, the short DNA sequences produced by modern sequencers require numerous quality control (QC) measures to reduce errors, remove technical contaminants, or merge paired-end reads together into longer or higher-quality contigs. Many tools for each step exist, but choosing the appropriate methods and usage parameters can be challenging because the parameterization of each step depends on the particularities of the sequencing technology used, the type of samples being analyzed, and the stochasticity of the instrumentation and sample preparation. Furthermore, end users may not know all of the relevant information about how their data were generated, such as the expected overlap for paired-end sequences or type of adaptors used to make informed choices. This increasing complexity and nuance demand a pipeline that combines existing steps together in a user-friendly way and, when possible, learns reasonable quality parameters from the data automatically. We propose a user-friendly quality control pipeline called SHI7 (canonically pronounced "shizen"), which aims to simplify quality control of short-read data for the end user by predicting presence and/or type of common sequencing adaptors, what quality scores to trim, whether the data set is shotgun or amplicon sequencing, whether reads are paired end or single end, and whether pairs are stitchable, including the expected amount of pair overlap. We hope that SHI7 will make it easier for all researchers, expert and novice alike, to follow reasonable practices for short-read data quality control. Quality control of high-throughput DNA sequencing data is an important but sometimes laborious task requiring background knowledge of the sequencing protocol used (such as adaptor type, sequencing technology, insert size/stitchability, paired-endedness, etc.). Quality control protocols typically require applying this background knowledge to selecting and executing numerous quality control steps with the appropriate parameters, which is especially difficult when working with public data or data from collaborators who use different protocols. We have created a streamlined quality control pipeline intended to substantially simplify the process of DNA quality control from raw machine output files to actionable sequence data. In contrast to other methods, our proposed pipeline is easy to install and use and attempts to learn the necessary parameters from the data automatically with a single command.
下一代测序技术对许多生物学学科都非常重要;然而,由于技术和生物学上的限制,现代测序仪产生的短DNA序列需要众多质量控制(QC)措施来减少错误、去除技术污染物,或将双端读数合并成更长或质量更高的重叠群。针对每个步骤都有许多工具,但选择合适的方法和使用参数可能具有挑战性,因为每个步骤的参数设置取决于所使用的测序技术的特殊性、被分析样本的类型以及仪器和样本制备的随机性。此外,终端用户可能并不了解有关其数据如何生成的所有相关信息,例如双端序列的预期重叠或用于做出明智选择的接头类型。这种日益增加的复杂性和细微差别需要一个以用户友好的方式将现有步骤组合在一起的流程,并且在可能的情况下,能从数据中自动学习合理的质量参数。我们提出了一个名为SHI7(标准发音为“shizen”)的用户友好型质量控制流程,其目的是通过预测常见测序接头的存在和/或类型、要修剪的质量分数、数据集是鸟枪法测序还是扩增子测序、读数是双端还是单端以及双端是否可拼接(包括预期的双端重叠量),来为终端用户简化短读数据的质量控制。我们希望SHI7能让所有研究人员,无论是专家还是新手,都更容易遵循短读数据质量控制的合理做法。高通量DNA测序数据的质量控制是一项重要但有时很费力的任务,需要对所使用的测序方案有背景知识(如接头类型、测序技术、插入片段大小/可拼接性、双端性等)。质量控制方案通常需要应用这些背景知识来选择并执行众多具有适当参数的质量控制步骤,在处理公共数据或来自使用不同方案的合作者的数据时尤其困难。我们创建了一个简化的质量控制流程,旨在从原始机器输出文件到可操作的序列数据,大幅简化DNA质量控制过程。与其他方法不同,我们提出的流程易于安装和使用,并尝试通过单个命令从数据中自动学习必要的参数。