Comeau André M, Douglas Gavin M, Langille Morgan G I
CGEB-Integrated Microbiome Resource (IMR) and Department of Pharmacology, Dalhousie University, Halifax, Canada.
mSystems. 2017 Jan 3;2(1). doi: 10.1128/mSystems.00127-16. eCollection 2017 Jan-Feb.
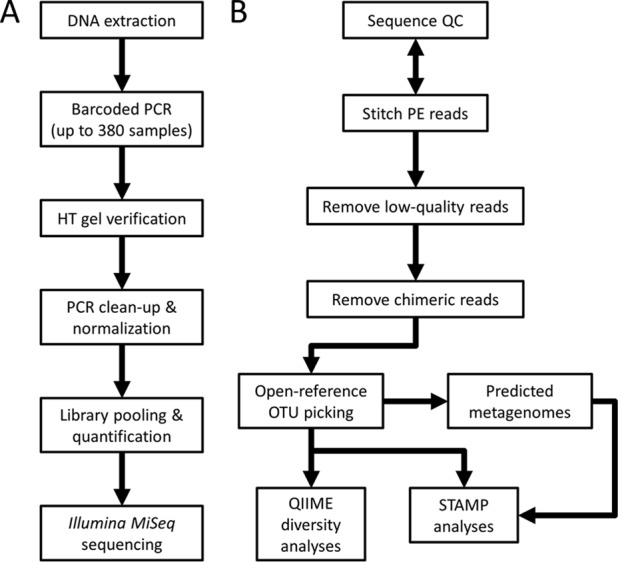
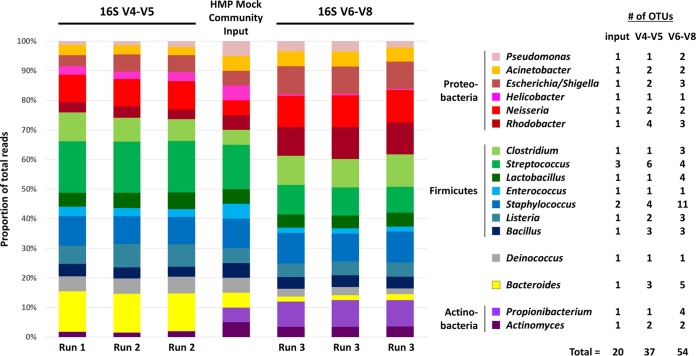
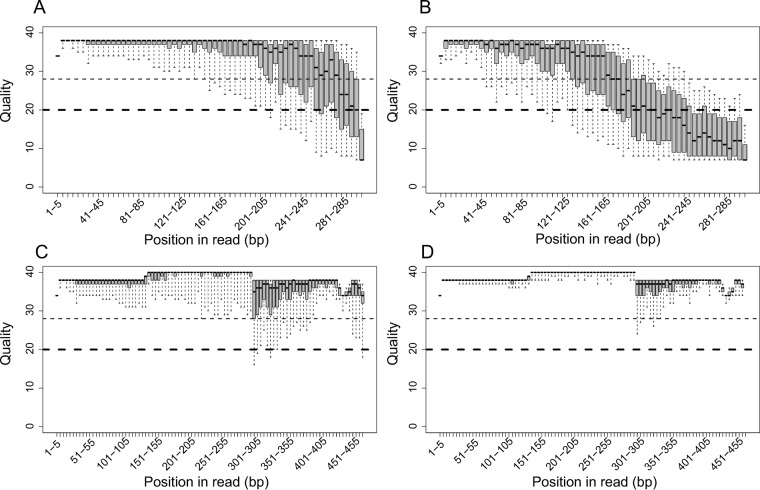
Sequence-based approaches to study microbiomes, such as 16S rRNA gene sequencing and metagenomics, are uncovering associations between microbial taxa and a myriad of factors. A drawback of these approaches is that the necessary sequencing library preparation and bioinformatic analyses are complicated and continuously changing, which can be a barrier for researchers new to the field. We present three essential components to conducting a microbiome experiment from start to finish: first, a simplified and step-by-step custom gene sequencing protocol that requires limited lab equipment, is cost-effective, and has been thoroughly tested and utilized on various sample types; second, a series of scripts to integrate various commonly used bioinformatic tools that is available as a standalone installation or as a single downloadable virtual image; and third, a set of bioinformatic workflows and tutorials to provide step-by-step guidance and education for those new to the microbiome field. This resource will provide the foundations for those newly entering the microbiome field and will provide much-needed guidance and best practices to ensure that quality microbiome research is undertaken. All protocols, scripts, workflows, tutorials, and virtual images are freely available through the Microbiome Helper website (https://github.com/mlangill/microbiome_helper/wiki). As the microbiome field continues to grow, a multitude of researchers are learning how to conduct proper microbiome experiments. We outline here a streamlined and custom approach to processing samples from detailed sequencing library construction to step-by-step bioinformatic standard operating procedures. This allows for rapid and reliable microbiome analysis, allowing researchers to focus more on their experiment design and results. Our sequencing protocols, bioinformatic tutorials, and bundled software are freely available through Microbiome Helper. As the microbiome research field continues to evolve, Microbiome Helper will be updated with new protocols, scripts, and training materials.
基于序列的微生物群落研究方法,如16S rRNA基因测序和宏基因组学,正在揭示微生物分类群与众多因素之间的关联。这些方法的一个缺点是,必要的测序文库制备和生物信息学分析复杂且不断变化,这可能成为该领域新手的障碍。我们介绍了从头到尾进行微生物群落实验的三个基本要素:第一,一个简化的、循序渐进的定制基因测序方案,该方案所需实验室设备有限,具有成本效益,并且已经在各种样本类型上进行了全面测试和应用;第二,一系列整合各种常用生物信息学工具的脚本,这些脚本可以单独安装,也可以作为单个可下载的虚拟镜像获取;第三,一套生物信息学工作流程和教程,为微生物群落领域的新手提供逐步指导和培训。该资源将为新进入微生物群落领域的人提供基础,并将提供急需的指导和最佳实践,以确保开展高质量的微生物群落研究。所有方案、脚本、工作流程、教程和虚拟镜像均可通过微生物群落助手网站(https://github.com/mlangill/microbiome_helper/wiki)免费获取。随着微生物群落领域的不断发展,众多研究人员正在学习如何进行适当的微生物群落实验。我们在此概述一种简化的定制方法,用于处理从详细的测序文库构建到逐步的生物信息学标准操作程序的样本。这允许进行快速可靠的微生物群落分析,使研究人员能够更多地关注他们的实验设计和结果。我们的测序方案、生物信息学教程和捆绑软件均可通过微生物群落助手免费获取。随着微生物群落研究领域的不断发展,微生物群落助手将更新新的方案、脚本和培训材料。