III rd Medical Department with Hematology, Medical Oncology, Hemostaseology, Infectious Diseases and Rheumatology, Oncologic Center; Salzburg Cancer Research Institute (SCRI) with Laboratory of Immunological and Molecular Cancer Research (LIMCR) and Center for Clinical Cancer and Immunology Trials (CCCIT); Paracelsus Medical University Salzburg, Salzburg, Austria.
Division of Bioinformatics, Biocenter, Medical University of Innsbruck, Innsbruck, Austria.
Theranostics. 2018 Mar 11;8(8):2278-2288. doi: 10.7150/thno.23544. eCollection 2018.
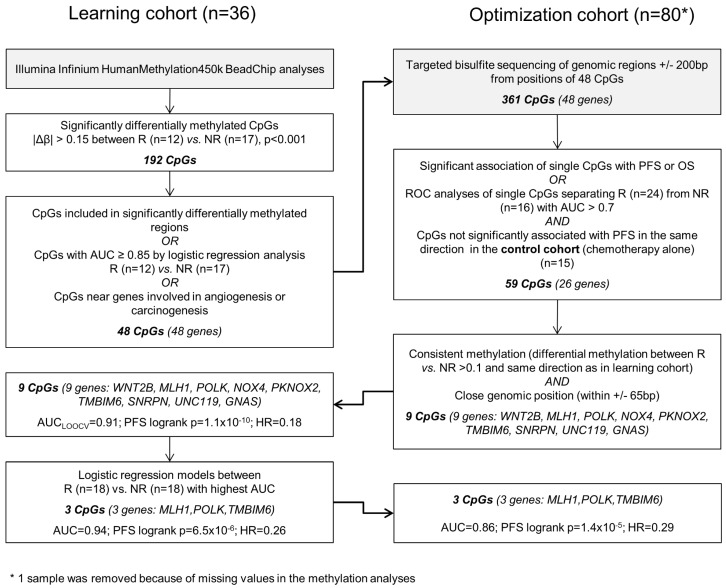
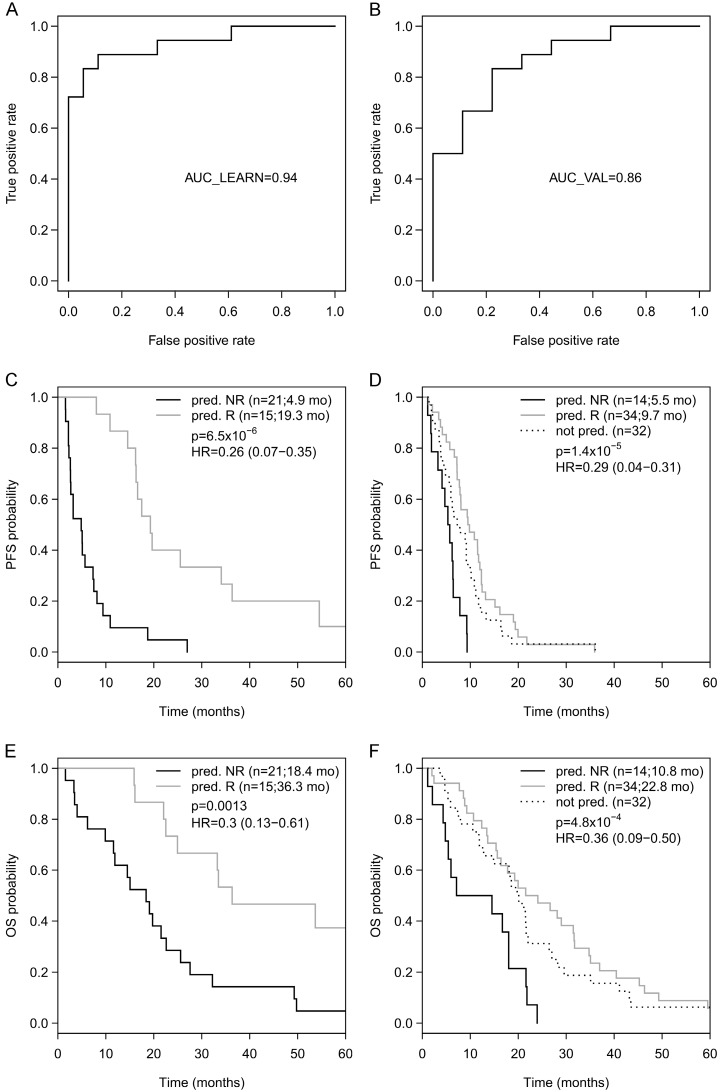
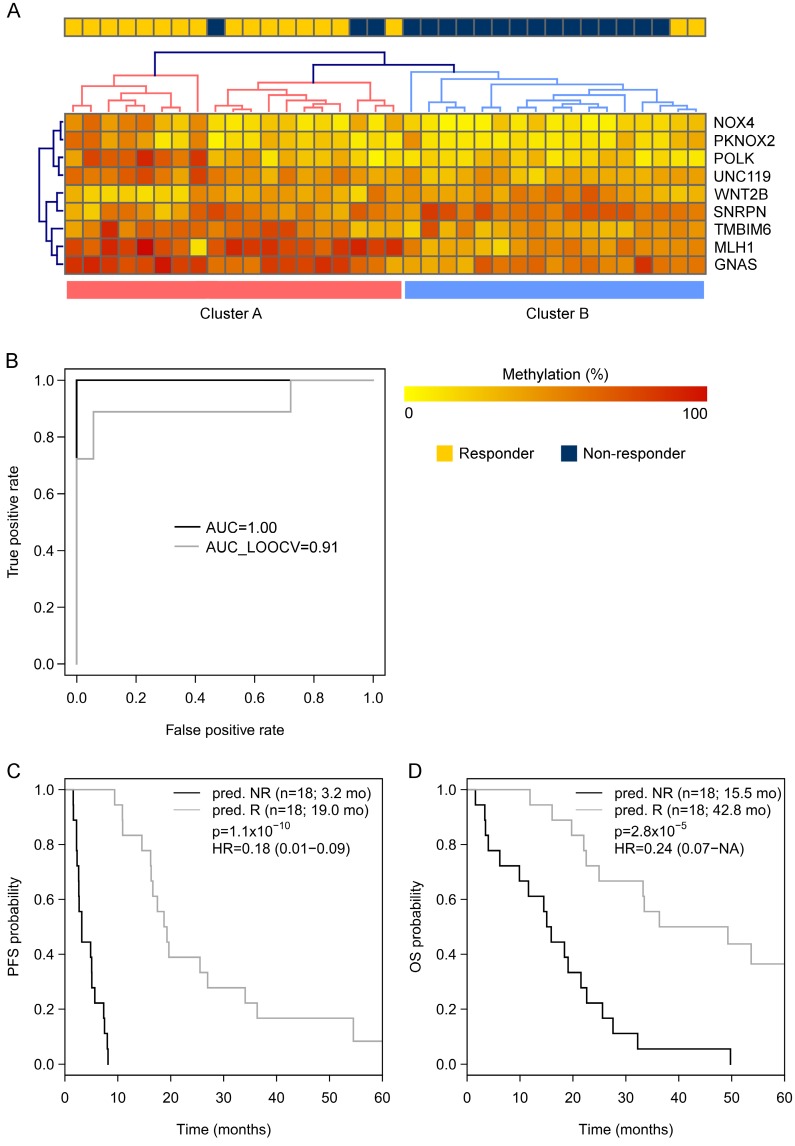
Biomarkers predicting response to bevacizumab in breast cancer are still missing. Since epigenetic modifications can contribute to an aberrant regulation of angiogenesis and treatment resistance, we investigated the influence of DNA methylation patterns on bevacizumab efficacy. Genome-wide methylation profiling using the was performed in archival FFPE specimens of 36 patients with HER2-negative metastatic breast cancer treated with chemotherapy in combination with bevacizumab as first-line therapy (). Based on objective response and progression-free survival (PFS) and considering ER expression, patients were divided in responders (R) and non-responders (NR). Significantly differentially methylated gene loci (CpGs) with a strong change in methylation levels (Δβ>0.15 or Δβ<-0.15) between R and NR were identified and further investigated in 80 bevacizumab-treated breast cancer patients () and in 15 patients treated with chemotherapy alone () using targeted deep amplicon bisulfite sequencing. Methylated gene loci were considered predictive if there was a significant association with outcome (PFS) in the but not in the using Spearman rank correlation, Cox regression, and logrank test. Differentially methylated loci in 48 genes were identified, allowing a good separation between R and NR (odds ratio (OR) 101, p<0.0001). Methylation of at least one cytosine in 26 gene-regions was significantly associated with progression-free survival (PFS) in the , but not in the . Using information from the , the panel was reduced to a 9-gene signature, which could divide patients from the into 2 clusters, thereby predicting response with an OR of 40 (<0.001) and an AUC of 0.91 (LOOCV). A further restricted 3-gene methylation model showed a significant association of with longer PFS in the and even in multivariate analysis with an excellent and good separation of R and NR with AUC=0.94 and AUC=0.86, respectively. Both a 9-gene and 3-gene methylation signature can discriminate between R and NR to a bevacizumab-based therapy in MBC and could help identify patients deriving greater benefit from bevacizumab.
用于预测乳腺癌对贝伐珠单抗反应的生物标志物仍然缺失。由于表观遗传修饰可能导致血管生成和治疗耐药性的异常调节,我们研究了 DNA 甲基化模式对贝伐珠单抗疗效的影响。
使用 对 36 例接受曲妥珠单抗联合化疗一线治疗的 HER2 阴性转移性乳腺癌患者的存档 FFPE 标本进行全基因组甲基化谱分析。基于客观缓解和无进展生存期(PFS),并考虑 ER 表达,将患者分为缓解者(R)和非缓解者(NR)。在 R 和 NR 之间,鉴定出具有强甲基化水平变化(Δβ>0.15 或 Δβ<-0.15)的差异甲基化基因座(CpG),并在 80 例接受贝伐珠单抗治疗的乳腺癌患者()和 15 例单独接受化疗的患者()中进一步进行靶向深度扩增子亚硫酸氢盐测序。如果在 中,与结局(PFS)存在显著关联(Spearman 秩相关、Cox 回归和对数秩检验),则认为甲基化基因座具有预测性。在 中则不然。
在 48 个基因中鉴定出差异甲基化基因座,能够很好地区分 R 和 NR(优势比(OR)101,p<0.0001)。至少一个基因座中 26 个基因的甲基化与 中的无进展生存期(PFS)显著相关,但在 中则不然。利用 中的信息,将该面板减少到 9 个基因的特征,该特征可将患者分为 2 个聚类,从而以 40(<0.001)的 OR 和 0.91(LOOCV)的 AUC 预测反应。进一步限制的 3 个基因甲基化模型显示,在 中与更长的 PFS 显著相关,甚至在多变量分析中,也能以 AUC=0.94 和 AUC=0.86 极好和良好地分离 R 和 NR。
9 个基因和 3 个基因甲基化特征均可区分贝伐珠单抗治疗转移性乳腺癌患者的反应,并有助于确定从贝伐珠单抗治疗中获益更大的患者。