Critical Care Medicine Department, NIH Clinical Center, National Institutes of Health, Bethesda, Maryland, USA
Critical Care Medicine Department, NIH Clinical Center, National Institutes of Health, Bethesda, Maryland, USA.
mBio. 2018 May 8;9(3):e00381-18. doi: 10.1128/mBio.00381-18.
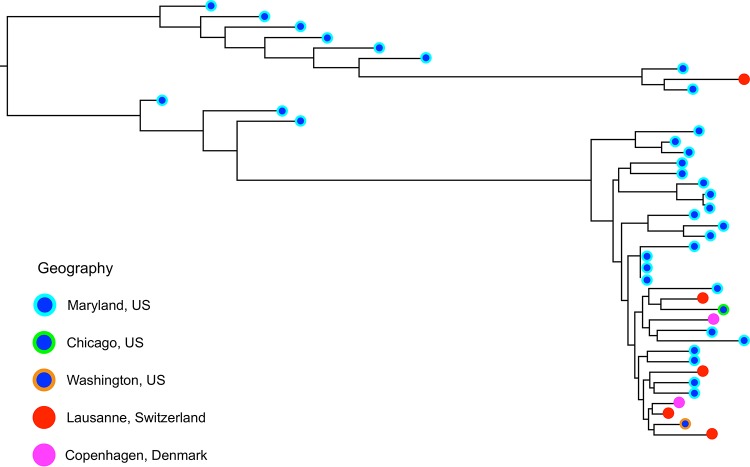
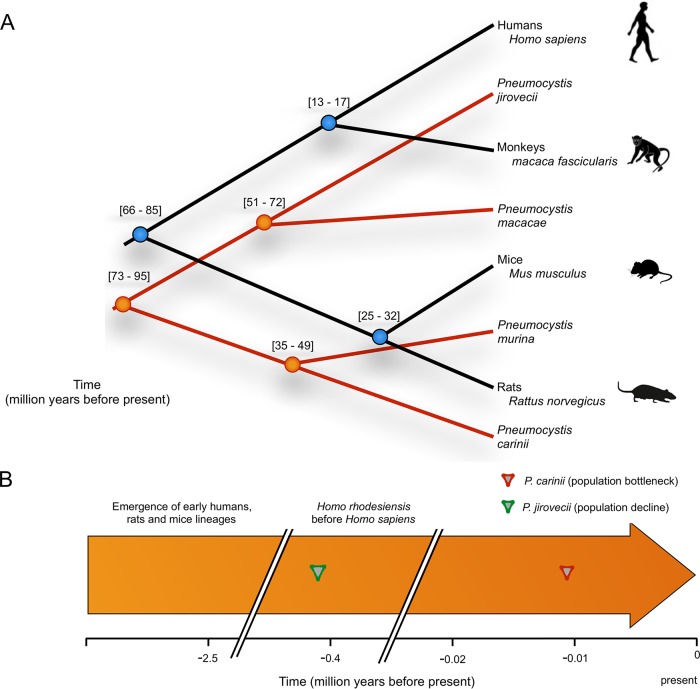
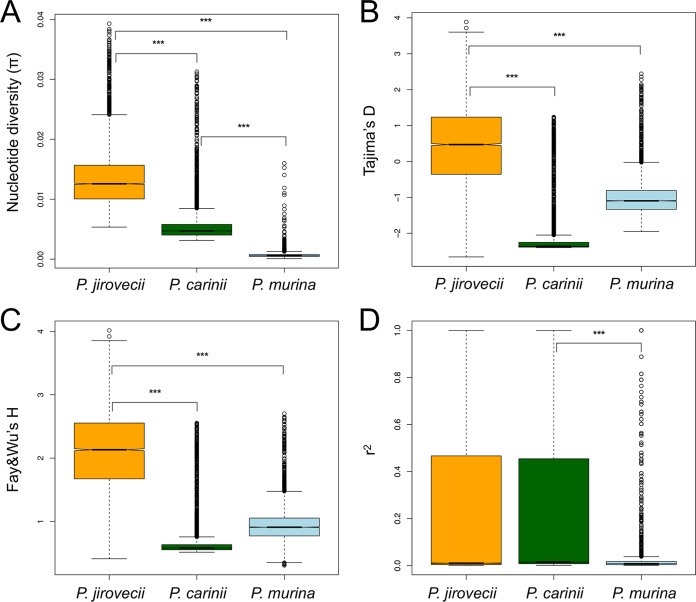
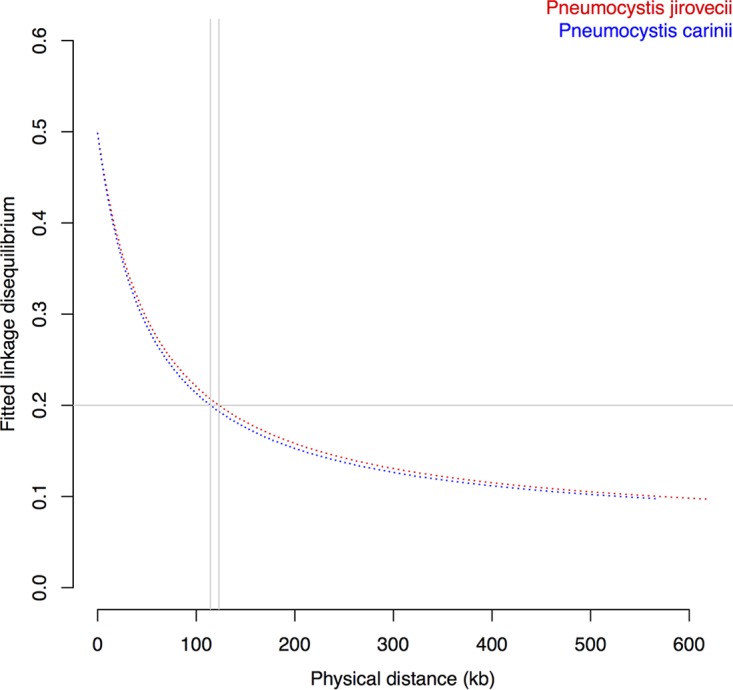
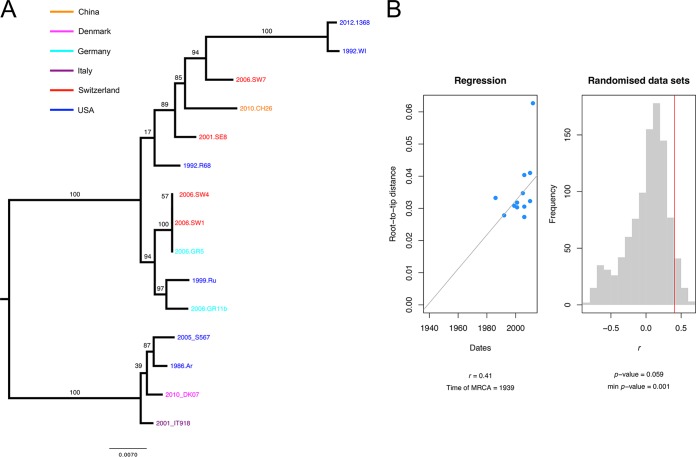
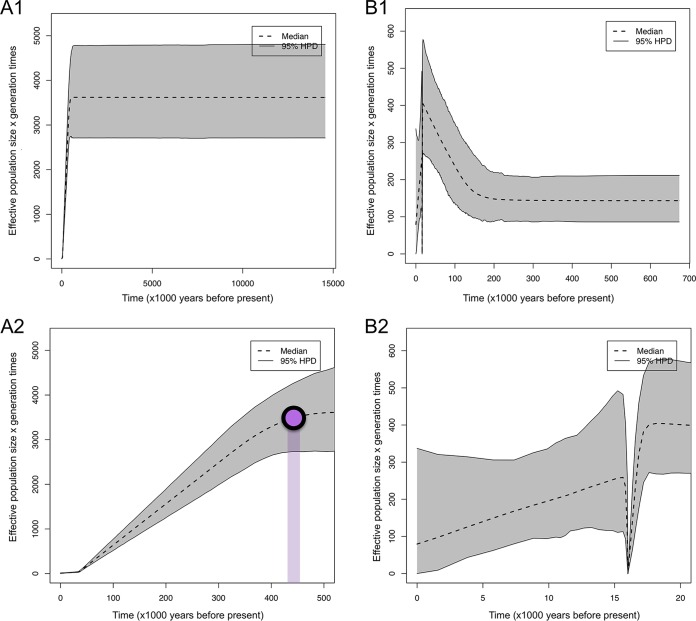
species are opportunistic mammalian pathogens that cause severe pneumonia in immunocompromised individuals. These fungi are highly host specific and uncultivable Human infections present major challenges because of a limited therapeutic arsenal and the rise of drug resistance. To investigate the diversity and demographic history of natural populations of infecting humans, rats, and mice, we performed whole-genome and large-scale multilocus sequencing of infected tissues collected in various geographic locations. Here, we detected reduced levels of recombination and variations in historical demography, which shape the global population structures. We report estimates of evolutionary rates, levels of genetic diversity, and population sizes. Molecular clock estimates indicate that species diverged before their hosts, while the asynchronous timing of population declines suggests host shifts. Our results have uncovered complex patterns of genetic variation influenced by multiple factors that shaped the adaptation of populations during their spread across mammals. Understanding how natural pathogen populations evolve and identifying the determinants of genetic variation are central issues in evolutionary biology. , a fungal pathogen which infects mammals exclusively, provides opportunities to explore these issues. In humans, can cause a life-threatening pneumonia in immunosuppressed individuals. In analysis of different species infecting humans, rats, and mice, we found that there are high infection rates and that natural populations maintain a high level of genetic variation despite low levels of recombination. We found no evidence of population structuring by geography. Our comparisons of the times of divergence of these species to their respective hosts suggest that may have undergone recent host shifts. The results demonstrate that strains are widely disseminated geographically and provide a new understanding of the evolution of these pathogens.
是一种机会主义的哺乳动物病原体,可导致免疫功能低下的个体发生严重肺炎。这些真菌具有高度的宿主特异性,且无法培养。由于治疗手段有限,以及耐药性的出现,人类感染这些真菌带来了重大挑战。为了研究感染人类、大鼠和小鼠的机会性病原体的多样性和种群历史,我们对从不同地理位置采集的感染组织进行了全基因组和大规模多位点测序。在这里,我们检测到重组水平降低和历史人口统计学的变化,这些变化塑造了全球种群结构。我们报告了进化率、遗传多样性水平和种群大小的估计值。分子钟估计表明,在其宿主之前就已经发生了物种分化,而种群下降的不同步时间表明发生了宿主转移。我们的研究结果揭示了受多种因素影响的复杂遗传变异模式,这些因素塑造了 种群在其在哺乳动物中传播过程中的适应性。了解自然病原体种群如何进化以及确定遗传变异的决定因素是进化生物学的核心问题。作为一种专门感染哺乳动物的真菌病原体,为探索这些问题提供了机会。在人类中, 可导致免疫抑制个体发生危及生命的肺炎。在对感染人类、大鼠和小鼠的不同 物种的分析中,我们发现尽管重组水平较低,但自然种群仍保持着高水平的遗传变异和高感染率。我们没有发现地理因素导致的种群结构的证据。我们将这些物种的分化时间与其各自的宿主进行比较,表明 可能发生了近期的宿主转移。研究结果表明, 菌株在地理上广泛传播,并为这些病原体的进化提供了新的认识。