Molecular and Cellular Oncology, ENT/University Hospital of Mainz, Mainz, 55131, Germany.
Institute for Medical Biostatistics, Epidemiology and Informatics (IMBEI), University of Mainz Medical Center, Mainz, 55101, Germany.
Sci Rep. 2018 May 9;8(1):7326. doi: 10.1038/s41598-018-25512-7.
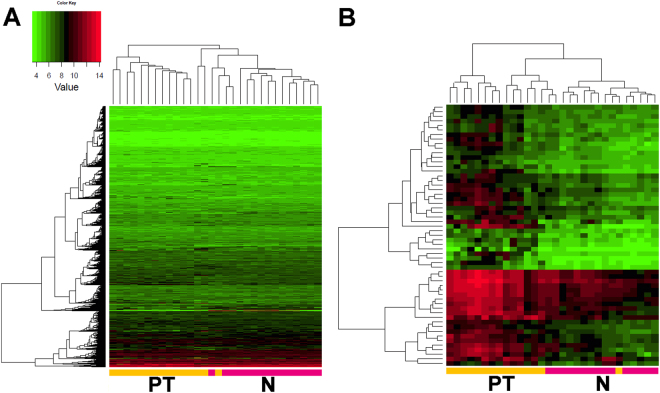
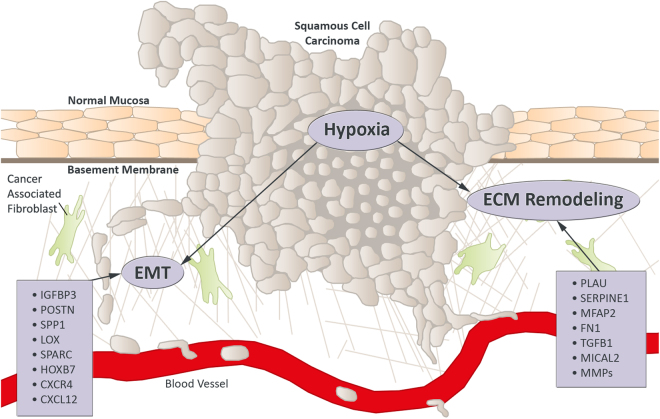
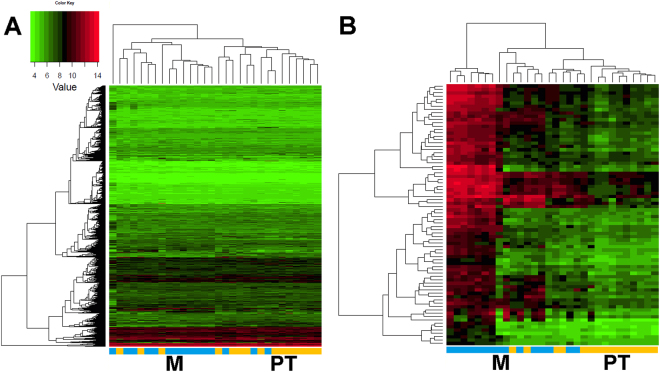
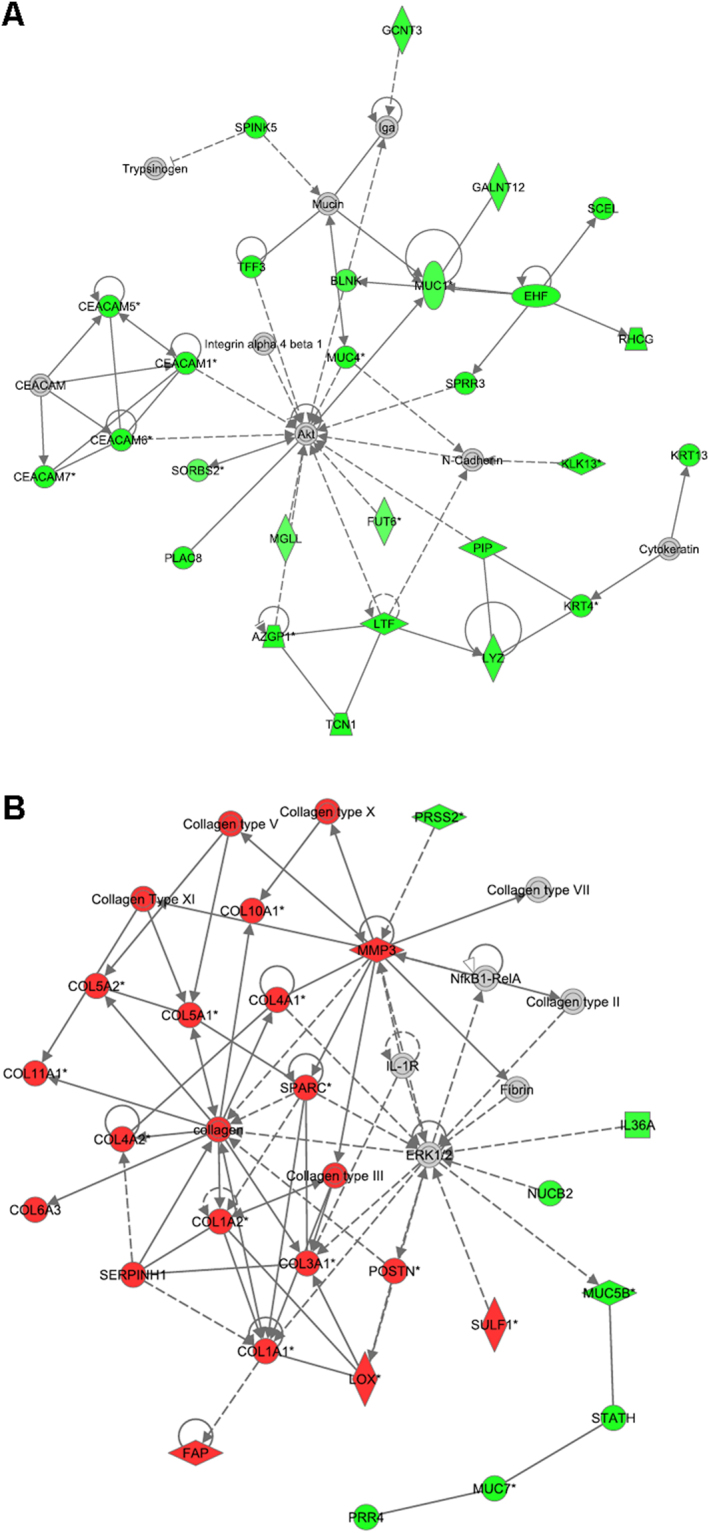
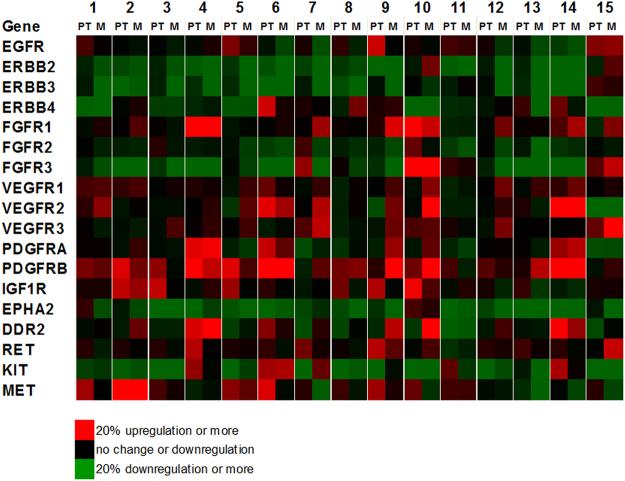
Head and neck squamous cell carcinoma (HNSCC) often metastasize to lymph nodes resulting in poor prognosis for patients. Unfortunately, the underlying molecular mechanisms contributing to tumour aggressiveness, recurrences, and metastasis are still not fully understood. However, such knowledge is key to identify biomarkers and drug targets to improve prognosis and treatments. Consequently, we performed genome-wide expression profiling of 15 primary HNSSCs compared to corresponding lymph node metastases and non-malignant tissue of the same patient. Differentially expressed genes were bioinformatically exploited applying stringent filter criteria, allowing the discrimination between normal mucosa, primary tumours, and metastases. Signalling networks involved in invasion contain remodelling of the extracellular matrix, hypoxia-induced transcriptional modulation, and the recruitment of cancer associated fibroblasts, ultimately converging into a broad activation of PI3K/AKT-signalling pathway in lymph node metastasis. Notably, when we compared the diagnostic and prognostic value of sequencing data with our expression analysis significant differences were uncovered concerning the expression of the receptor tyrosine kinases EGFR and ERBB2, as well as other oncogenic regulators. Particularly, upregulated receptor tyrosine kinase combinations for individual patients varied, implying potential compensatory and resistance mechanisms against specific targeted therapies. Collectively, we here provide unique transcriptional profiles for disease predictions and comprehensively analyse involved signalling pathways in advanced HNSCC.
头颈部鳞状细胞癌(HNSCC)常发生淋巴结转移,导致患者预后不良。不幸的是,导致肿瘤侵袭性、复发和转移的潜在分子机制仍未完全阐明。然而,这些知识是识别生物标志物和药物靶点以改善预后和治疗的关键。因此,我们对 15 例原发 HNSCC 与相应的淋巴结转移和同一患者的非恶性组织进行了全基因组表达谱分析。应用严格的筛选标准,对差异表达基因进行生物信息学挖掘,能够区分正常黏膜、原发肿瘤和转移。参与侵袭的信号网络包含细胞外基质的重塑、缺氧诱导的转录调节以及癌相关成纤维细胞的募集,最终导致淋巴结转移中广泛激活 PI3K/AKT 信号通路。值得注意的是,当我们将测序数据的诊断和预后价值与我们的表达分析进行比较时,发现 EGFR 和 ERBB2 等受体酪氨酸激酶以及其他致癌调节剂的表达存在显著差异。特别是,个别患者上调的受体酪氨酸激酶组合不同,意味着针对特定靶向治疗存在潜在的补偿和耐药机制。总之,我们在这里提供了用于疾病预测的独特转录谱,并全面分析了晚期 HNSCC 中涉及的信号通路。