Jorge Paula, Garcia Elsa, Gonçalves Ana, Marques Isabel, Maia Nuno, Rodrigues Bárbara, Santos Helena, Fonseca Jacinta, Soares Gabriela, Correia Cecília, Reis-Lima Margarida, Cirigliano Vincenzo, Santos Rosário
Centro de Genética Médica Jacinto de Magalhães (CGMJM), Centro Hospitalar do Porto, CHP, E.P.E., Praça Pedro Nunes, 88 4099-028, Porto, Portugal.
Unit for Multidisciplinary Research in Biomedicine, Abel Salazar Institute of Biomedical Sciences, University of Porto - UMIB-ICBAS-UP, Porto, Portugal.
BMC Med Genet. 2018 May 10;19(1):74. doi: 10.1186/s12881-018-0589-6.
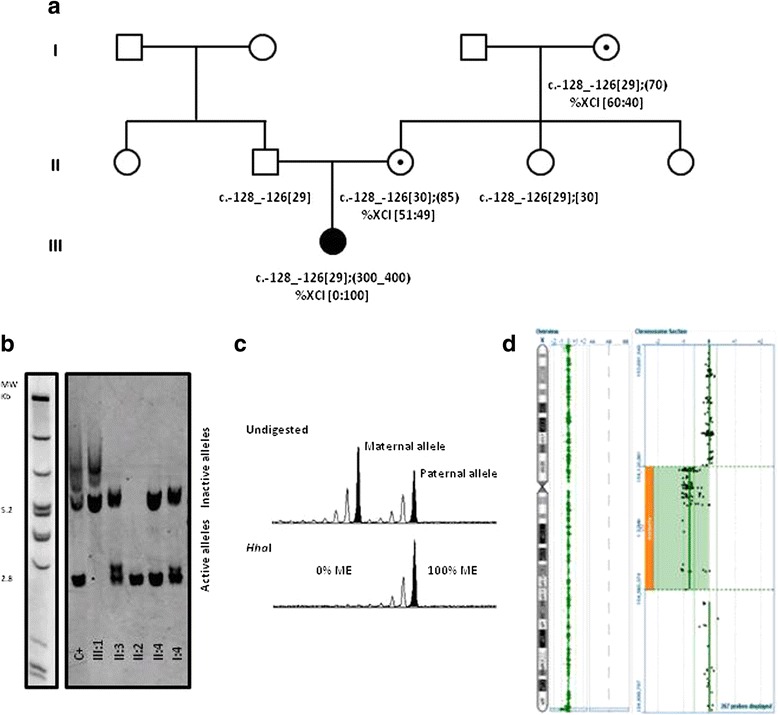
We describe a female infant with Fragile-X syndrome, with a fully expanded FMR1 allele and preferential inactivation of the homologous X-chromosome carrying a de novo deletion. This unusual and rare case demonstrates the importance of a detailed genomic approach, the absence of which could be misguiding, and calls for reflection on the current clinical and diagnostic workup for developmental disabilities.
We present a female infant, referred for genetic testing due to psychomotor developmental delay without specific dysmorphic features or relevant family history. FMR1 mutation screening revealed a methylated full mutation and a normal but inactive FMR1 allele, which led to further investigation. Complete skewing of X-chromosome inactivation towards the paternally-inherited normal-sized FMR1 allele was found. No pathogenic variants were identified in the XIST promoter. Microarray analysis revealed a 439 kb deletion at Xq28, in a region known to be associated with extreme skewing of X-chromosome inactivation.
Overall results enable us to conclude that the developmental delay is the cumulative result of a methylated FMR1 full mutation on the active X-chromosome and the inactivation of the other homologue carrying the de novo 439 kb deletion. Our findings should be taken into consideration in future guidelines for the diagnostic workup on the diagnosis of intellectual disabilities, particularly in female infant cases.
我们描述了一名患有脆性X综合征的女婴,其FMR1等位基因完全扩增,并且携带新发缺失的同源X染色体优先失活。这个不寻常且罕见的病例证明了详细基因组方法的重要性,缺乏该方法可能会产生误导,并促使人们对目前发育障碍的临床和诊断检查进行反思。
我们介绍了一名女婴,因精神运动发育迟缓而接受基因检测,该女婴没有特定的畸形特征或相关家族史。FMR1突变筛查显示一个甲基化的全突变和一个正常但无活性的FMR1等位基因,这促使进行进一步调查。发现X染色体失活完全偏向父系遗传的正常大小的FMR1等位基因。在XIST启动子中未发现致病变体。微阵列分析显示在Xq28处有一个439 kb的缺失,该区域已知与X染色体失活的极端偏向有关。
总体结果使我们能够得出结论,发育迟缓是活性X染色体上甲基化的FMR1全突变以及携带新发439 kb缺失的另一条同源染色体失活的累积结果。我们的发现应在未来智力残疾诊断检查的指南中予以考虑,尤其是在女婴病例中。