AstraZeneca, Oncology, IMED Biotech Unit, 1 Francis Crick Avenue, Cambridge, CB2 0RE, UK.
Institute of Organic Chemistry, University of Vienna, Währinger Straße 38, 1090, Vienna, Austria.
Angew Chem Int Ed Engl. 2018 Aug 13;57(33):10737-10741. doi: 10.1002/anie.201804551. Epub 2018 Jun 22.
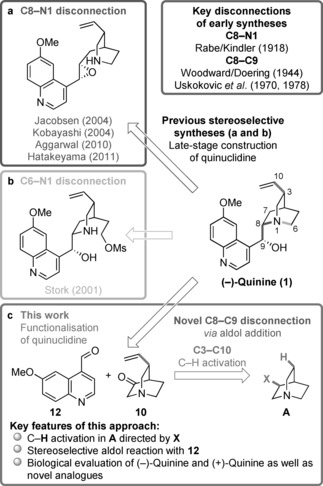
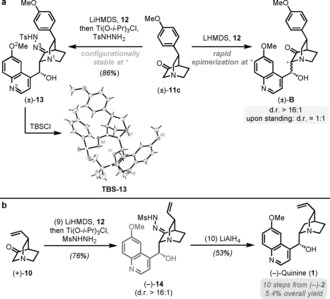
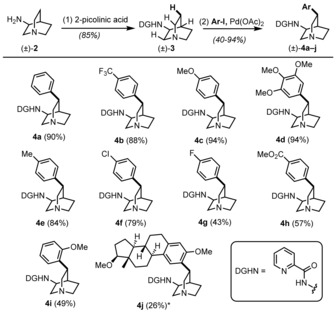
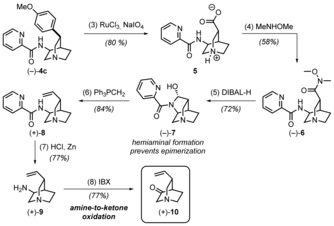
We report a novel approach to the classical natural product quinine that is based on two stereoselective key steps, namely a C-H activation and an aldol reaction, to unite the two heterocyclic moieties of the target molecule. This straightforward and flexible strategy enables a concise synthesis of natural (-)-quinine, the first synthesis of unnatural (+)-quinine, and also provides access to unprecedented C3-aryl analogues, which were prepared in only six steps. We additionally demonstrate that these structural analogues exhibit improved antimalarial activity compared with (-)-quinine both in vitro and in mice infected with Plasmodium berghei.
我们报告了一种新颖的方法来合成经典的天然产物奎宁,该方法基于两个立体选择性的关键步骤,即 C-H 活化和羟醛反应,以将目标分子的两个杂环部分连接起来。这种直接而灵活的策略使得简洁地合成天然(-)-奎宁、首次合成非天然(+)-奎宁以及获得前所未有的 C3-芳基类似物成为可能,这些类似物仅通过六步反应即可制备。我们还证明,与(-)-奎宁相比,这些结构类似物在体外和感染疟原虫(Plasmodium berghei)的小鼠中均表现出更好的抗疟活性。