Protein Biochemistry, Institute for Biochemistry, Freie Universität Berlin, Berlin, Germany.
Computational Molecular Biology Group, Institute for Mathematics, Freie Universität Berlin, Berlin, Germany.
Front Immunol. 2018 May 3;9:872. doi: 10.3389/fimmu.2018.00872. eCollection 2018.
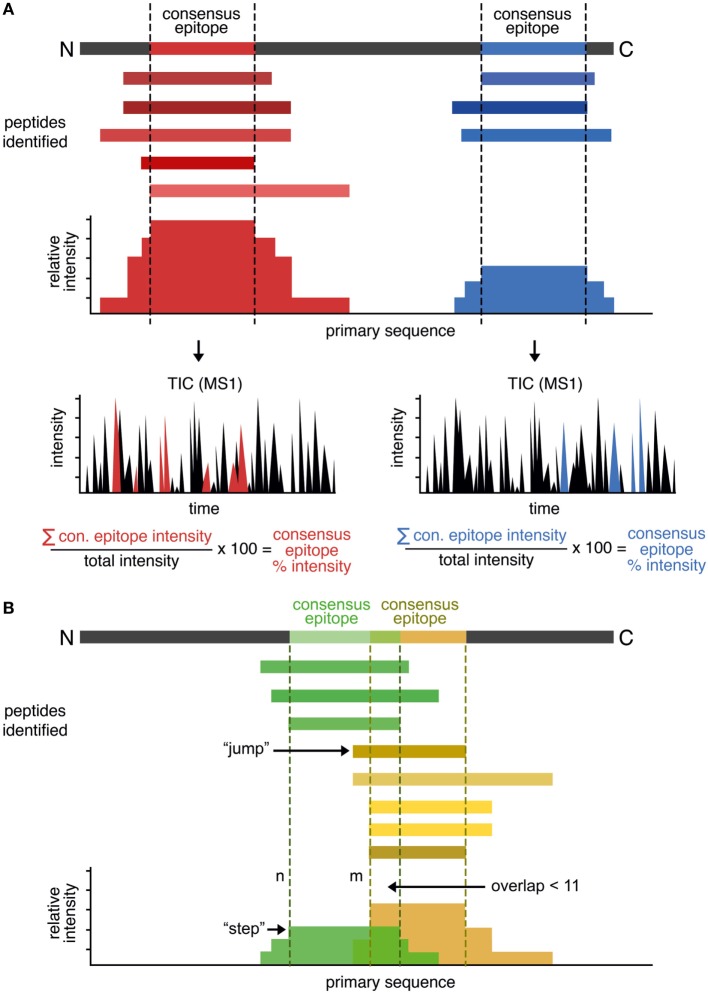
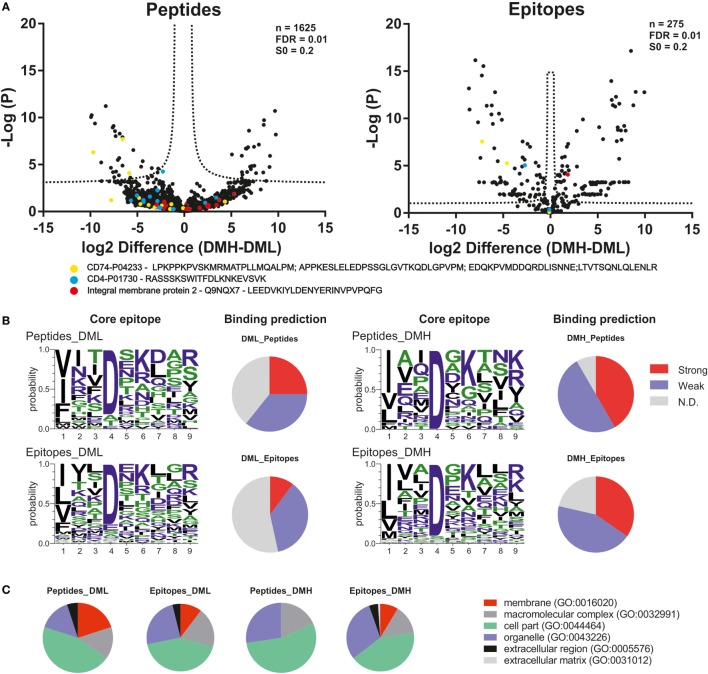
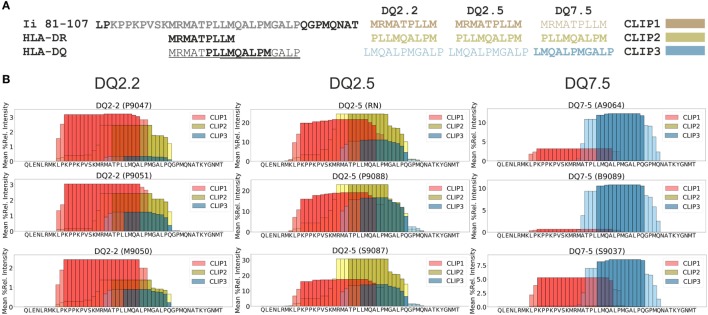
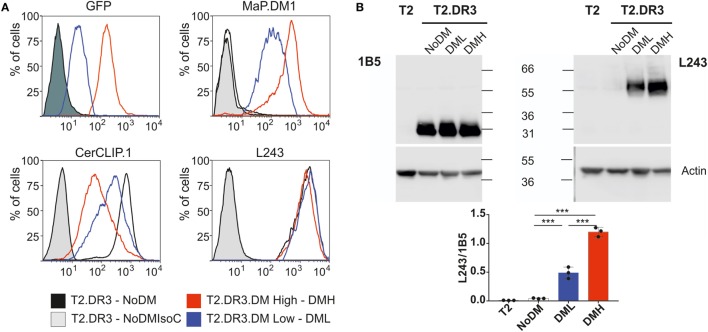
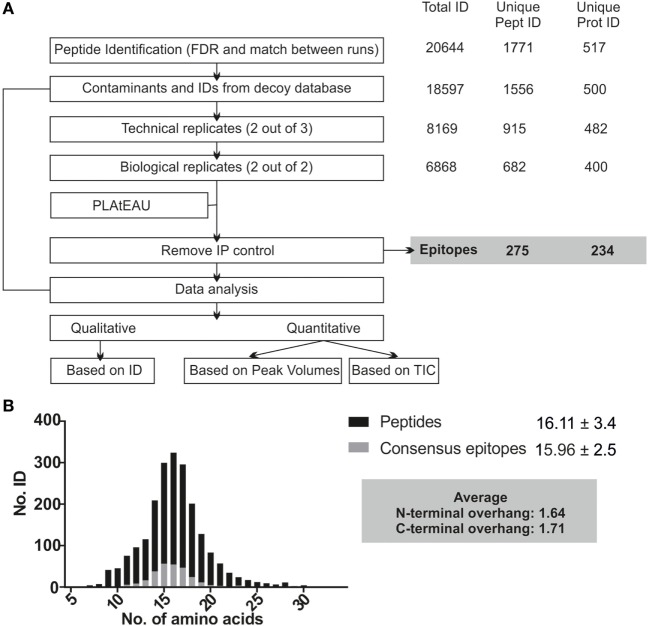
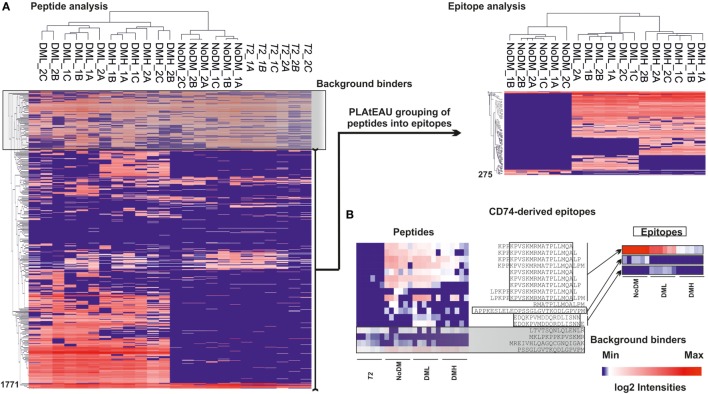
The major histocompatibility complex of class II (MHCII) immunopeptidome represents the repertoire of antigenic peptides with the potential to activate CD4 T cells. An understanding of how the relative abundance of specific antigenic epitopes affects the outcome of T cell responses is an important aspect of adaptive immunity and offers a venue to more rationally tailor T cell activation in the context of disease. Recent advances in mass spectrometric instrumentation, computational power, labeling strategies, and software analysis have enabled an increasing number of stratified studies on HLA ligandomes, in the context of both basic and translational research. A key challenge in the case of MHCII immunopeptidomes, often determined for different samples at distinct conditions, is to derive quantitative information on consensus epitopes from antigenic peptides of variable lengths. Here, we present the design and benchmarking of a new algorithm [peptide landscape antigenic epitope alignment utility (PLAtEAU)] allowing the identification and label-free quantification (LFQ) of shared consensus epitopes arising from series of nested peptides. The algorithm simplifies the complexity of the dataset while allowing the identification of nested peptides within relatively short segments of protein sequences. Moreover, we apply this algorithm to the comparison of the ligandomes of cell lines with two different expression levels of the peptide-exchange catalyst HLA-DM. Direct comparison of LFQ intensities determined at the peptide level is inconclusive, as most of the peptides are not significantly enriched due to poor sampling. Applying the PLAtEAU algorithm for grouping of the peptides into consensus epitopes shows that more than half of the total number of epitopes is preferentially and significantly enriched for each condition. This simplification and deconvolution of the complex and ambiguous peptide-level dataset highlights the value of the PLAtEAU algorithm in facilitating robust and accessible quantitative analysis of immunopeptidomes across cellular contexts. analysis of the peptides enriched for each HLA-DM expression conditions suggests a higher affinity of the pool of peptides isolated from the high DM expression samples. Interestingly, our analysis reveals that while for certain autoimmune-relevant epitopes their presentation increases upon DM expression others are clearly edited out from the peptidome.
主要组织相容性复合体 II(MHCII)免疫肽组代表了具有激活 CD4 T 细胞潜力的抗原肽库。了解特定抗原表位的相对丰度如何影响 T 细胞反应的结果是适应性免疫的一个重要方面,并为在疾病背景下更合理地调整 T 细胞激活提供了途径。质谱仪器、计算能力、标记策略和软件分析的最新进展使得 HLA 配体组在基础和转化研究方面的分层研究数量不断增加。在 MHCII 免疫肽组的情况下,一个关键挑战是从长度可变的抗原肽中得出共识表位的定量信息,这些免疫肽组通常是针对不同条件下的不同样本确定的。在这里,我们提出了一种新算法(肽景观抗原表位对齐实用程序(PLAtEU))的设计和基准测试,该算法允许从一系列嵌套肽中识别和无标记定量(LFQ)共享共识表位。该算法简化了数据集的复杂性,同时允许在相对较短的蛋白质序列段内识别嵌套肽。此外,我们将该算法应用于比较具有两种不同肽交换催化剂 HLA-DM 表达水平的细胞系的配体组。在肽水平上直接比较 LFQ 强度的结果是不确定的,因为由于采样不佳,大多数肽没有显著富集。应用 PLAtEU 算法将肽分组为共识表位表明,超过一半的总表位数对于每种条件都是优先且显著富集的。这种对复杂且模糊的肽水平数据集的简化和解卷积突出了 PLAtEU 算法在促进跨细胞背景下免疫肽组的稳健和可访问的定量分析方面的价值。对每种 HLA-DM 表达条件下富集的肽进行分析表明,从高 DM 表达样本中分离的肽池中肽的亲和力更高。有趣的是,我们的分析表明,虽然某些与自身免疫相关的表位在 DM 表达时会增加,但其他表位显然已从肽组中编辑掉了。